Chapter 4 Human PBMC with surface proteins (10X Genomics)
4.1 Introduction
Here, we describe a brief analysis of yet another peripheral blood mononuclear cell (PBMC) dataset from 10X Genomics (Zheng et al. 2017). Data are publicly available from the 10X Genomics website, from which we download the filtered gene/barcode count matrices for gene expression and cell surface proteins.
4.2 Data loading
library(BiocFileCache)
bfc <- BiocFileCache(ask=FALSE)
exprs.data <- bfcrpath(bfc, file.path(
"http://cf.10xgenomics.com/samples/cell-vdj/3.1.0",
"vdj_v1_hs_pbmc3",
"vdj_v1_hs_pbmc3_filtered_feature_bc_matrix.tar.gz"))
untar(exprs.data, exdir=tempdir())
library(DropletUtils)
sce.pbmc <- read10xCounts(file.path(tempdir(), "filtered_feature_bc_matrix"))
sce.pbmc <- splitAltExps(sce.pbmc, rowData(sce.pbmc)$Type)4.3 Quality control
We discard cells with high mitochondrial proportions and few detectable ADT counts.
library(scater)
is.mito <- grep("^MT-", rowData(sce.pbmc)$Symbol)
stats <- perCellQCMetrics(sce.pbmc, subsets=list(Mito=is.mito))
high.mito <- isOutlier(stats$subsets_Mito_percent, type="higher")
low.adt <- stats$`altexps_Antibody Capture_detected` < nrow(altExp(sce.pbmc))/2
discard <- high.mito | low.adt
sce.pbmc <- sce.pbmc[,!discard]We examine some of the statistics:
## Mode FALSE TRUE
## logical 6660 571## Mode FALSE
## logical 7231## Mode FALSE TRUE
## logical 6660 571We examine the distribution of each QC metric (Figure 4.1).
colData(unfiltered) <- cbind(colData(unfiltered), stats)
unfiltered$discard <- discard
gridExtra::grid.arrange(
plotColData(unfiltered, y="sum", colour_by="discard") +
scale_y_log10() + ggtitle("Total count"),
plotColData(unfiltered, y="detected", colour_by="discard") +
scale_y_log10() + ggtitle("Detected features"),
plotColData(unfiltered, y="subsets_Mito_percent",
colour_by="discard") + ggtitle("Mito percent"),
plotColData(unfiltered, y="altexps_Antibody Capture_detected",
colour_by="discard") + ggtitle("ADT detected"),
ncol=2
)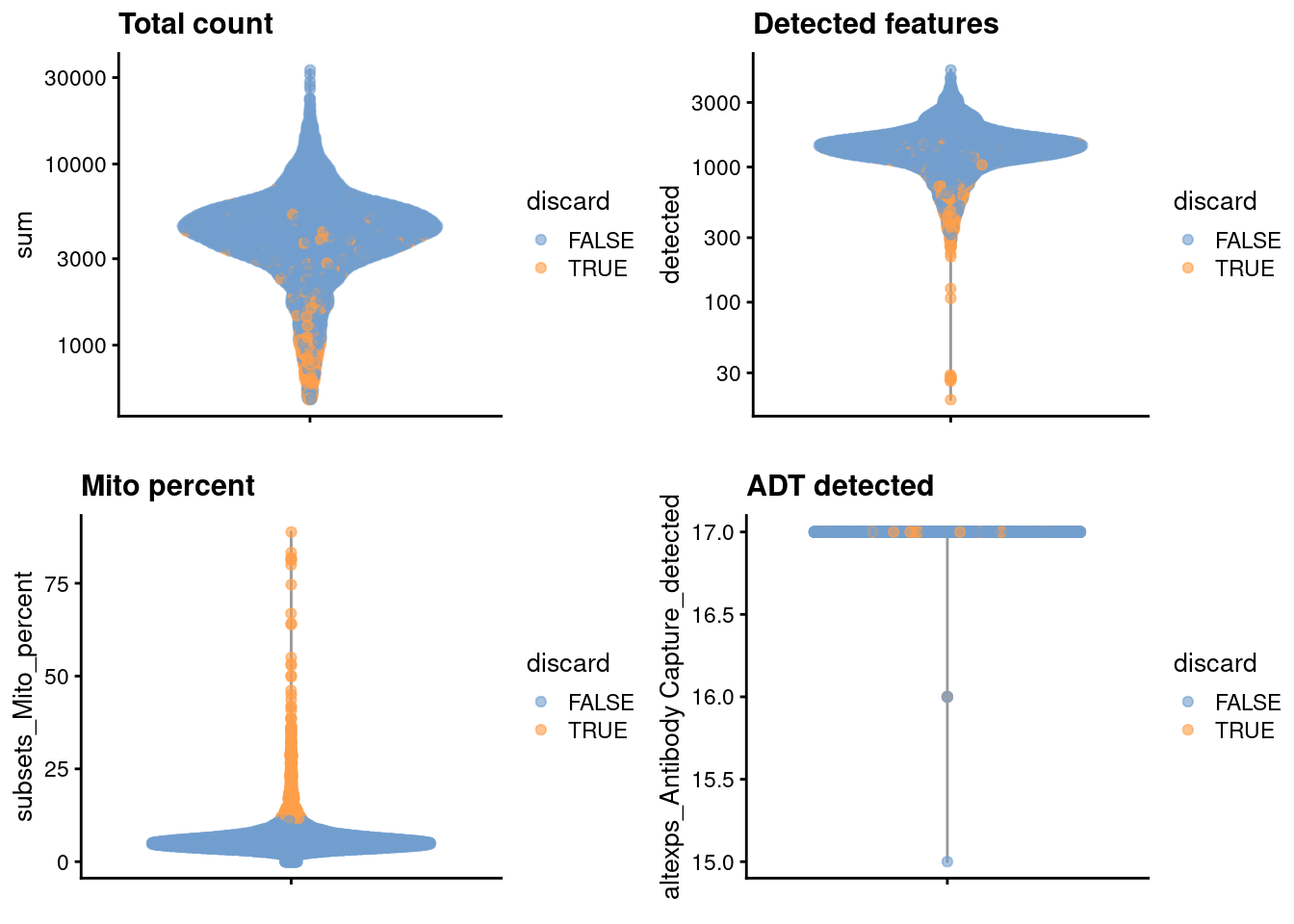
Figure 4.1: Distribution of each QC metric in the PBMC dataset, where each point is a cell and is colored by whether or not it was discarded by the outlier-based QC approach.
We also plot the mitochondrial proportion against the total count for each cell, as one does (Figure 4.2).
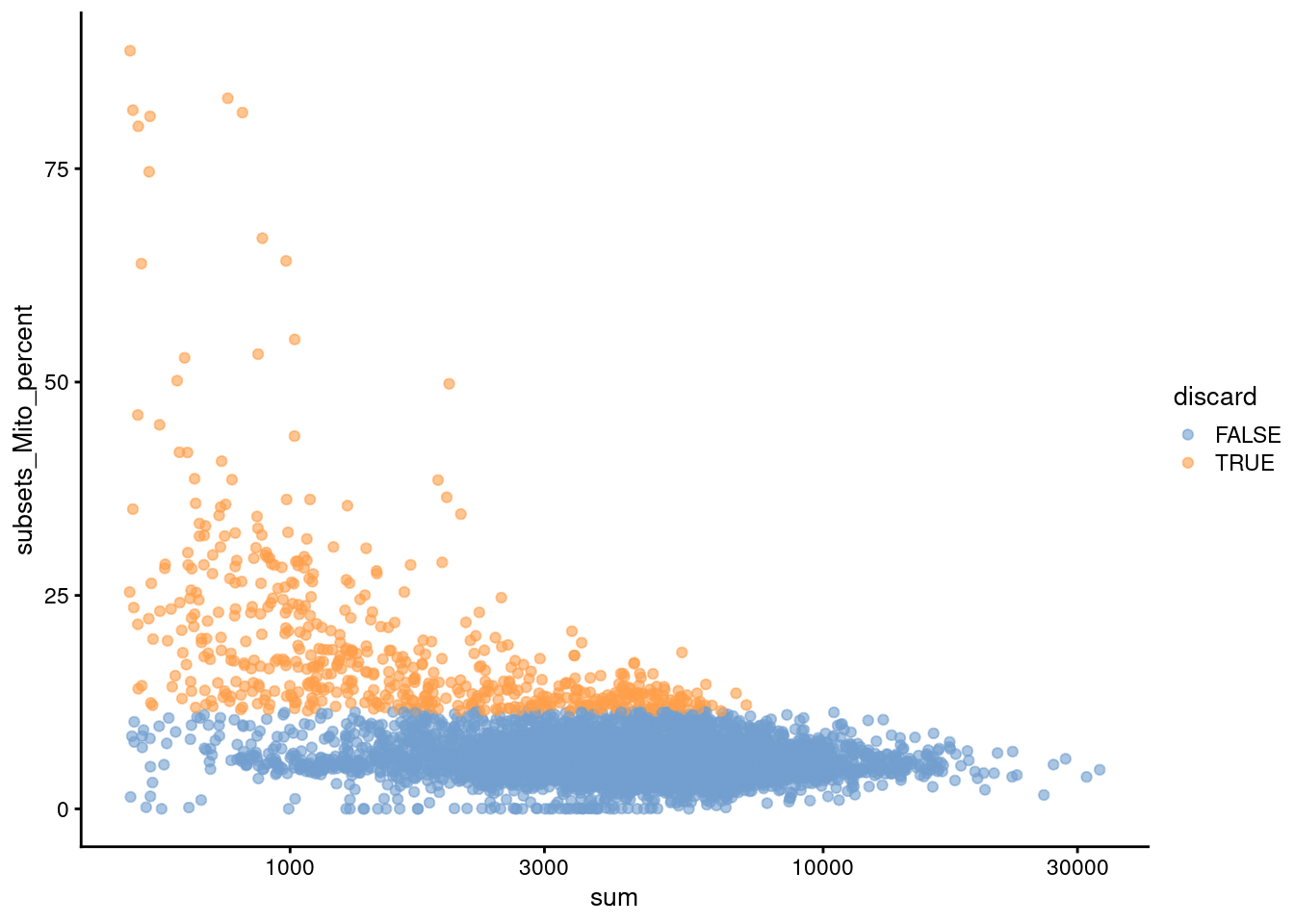
Figure 4.2: Percentage of UMIs mapped to mitochondrial genes against the totalcount for each cell.
4.4 Normalization
Computing size factors for the gene expression and ADT counts.
library(scran)
set.seed(1000)
clusters <- quickCluster(sce.pbmc)
sce.pbmc <- computeSumFactors(sce.pbmc, cluster=clusters)
altExp(sce.pbmc) <- computeMedianFactors(altExp(sce.pbmc))
sce.pbmc <- applySCE(sce.pbmc, logNormCounts)We generate some summary statistics for both sets of size factors:
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.074 0.717 0.909 1.000 1.128 9.100## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.10 0.70 0.83 1.00 1.03 227.36We also look at the distribution of size factors compared to the library size for each set of features (Figure 4.3).
par(mfrow=c(1,2))
plot(librarySizeFactors(sce.pbmc), sizeFactors(sce.pbmc), pch=16,
xlab="Library size factors", ylab="Deconvolution factors",
main="Gene expression", log="xy")
plot(librarySizeFactors(altExp(sce.pbmc)), sizeFactors(altExp(sce.pbmc)), pch=16,
xlab="Library size factors", ylab="Median-based factors",
main="Antibody capture", log="xy")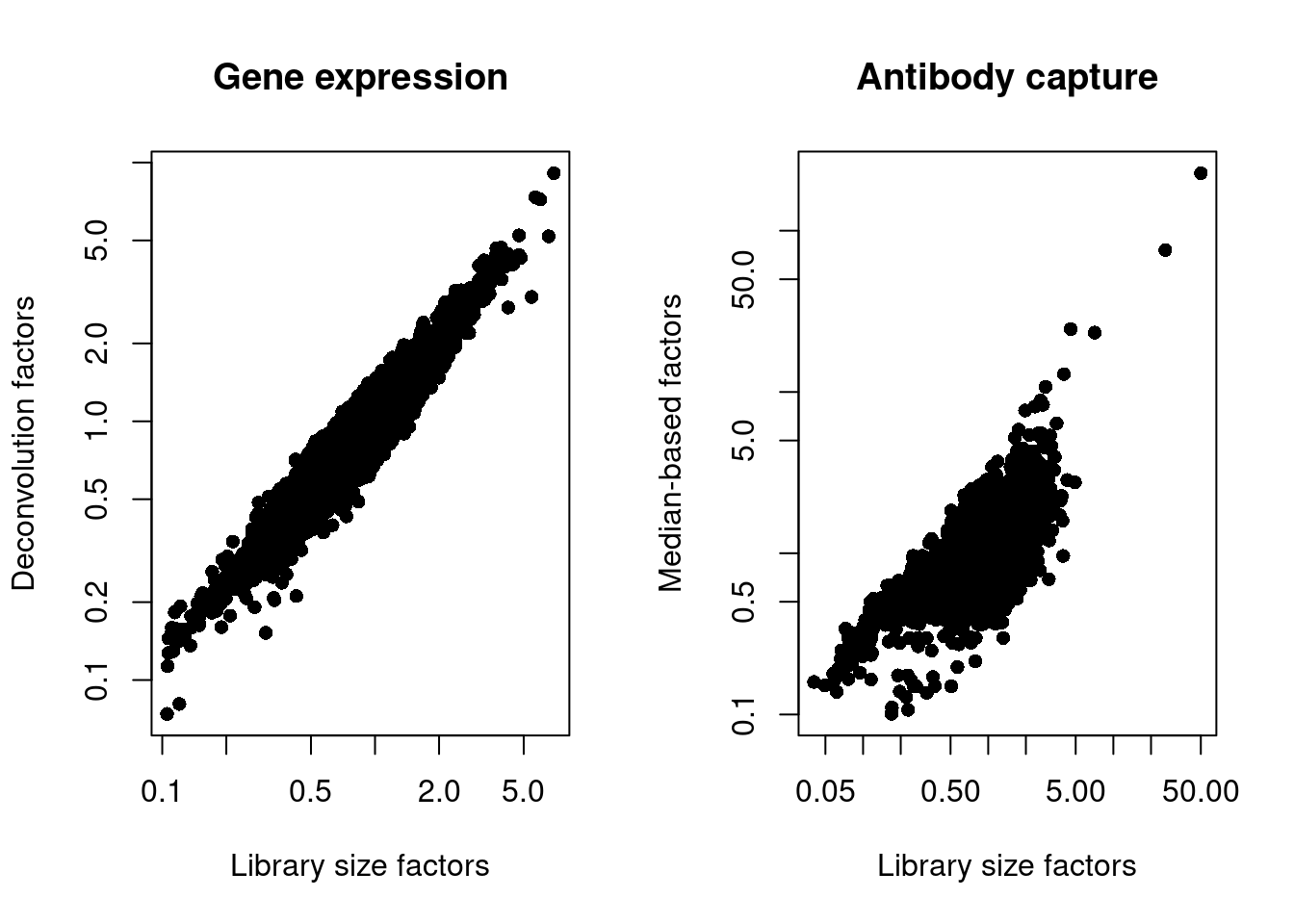
Figure 4.3: Plot of the deconvolution size factors for the gene expression values (left) or the median-based size factors for the ADT expression values (right) compared to the library size-derived factors for the corresponding set of features. Each point represents a cell.
4.5 Dimensionality reduction
We omit the PCA step for the ADT expression matrix, given that it is already so low-dimensional, and progress directly to \(t\)-SNE and UMAP visualizations.
4.6 Clustering
We perform graph-based clustering on the ADT data and use the assignments as the column labels of the alternative Experiment.
g.adt <- buildSNNGraph(altExp(sce.pbmc), k=10, d=NA)
clust.adt <- igraph::cluster_walktrap(g.adt)$membership
colLabels(altExp(sce.pbmc)) <- factor(clust.adt)We examine some basic statistics about the size of each cluster, their separation (Figure 4.4) and their distribution in our \(t\)-SNE plot (Figure 4.5).
##
## 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
## 160 507 662 39 691 1415 32 650 76 1037 121 47 68 25 15 562
## 17 18 19 20 21 22 23 24
## 139 32 44 120 84 65 52 17library(bluster)
mod <- pairwiseModularity(g.adt, clust.adt, as.ratio=TRUE)
library(pheatmap)
pheatmap::pheatmap(log10(mod + 10), cluster_row=FALSE, cluster_col=FALSE,
color=colorRampPalette(c("white", "blue"))(101))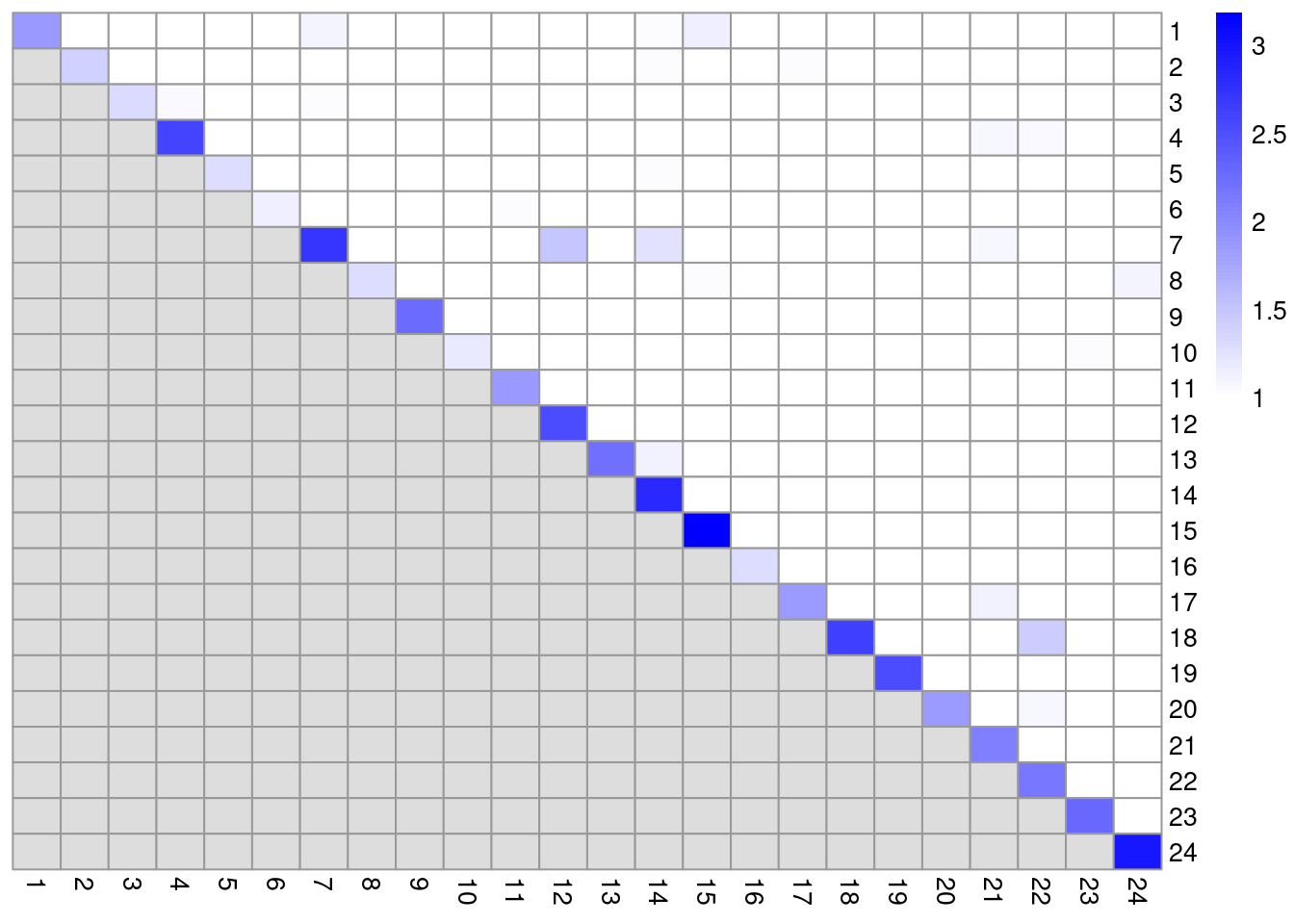
Figure 4.4: Heatmap of the pairwise cluster modularity scores in the PBMC dataset, computed based on the shared nearest neighbor graph derived from the ADT expression values.
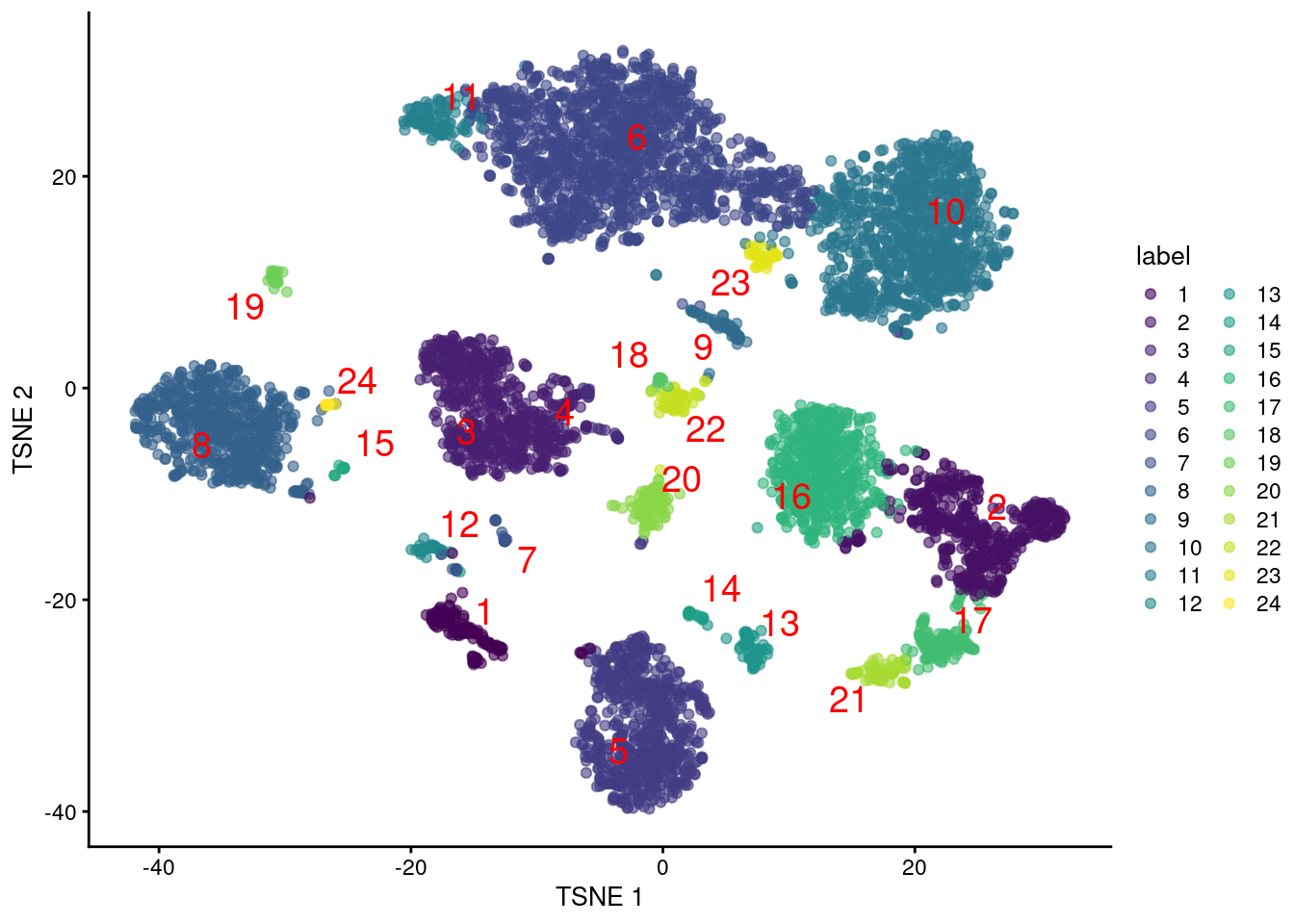
Figure 4.5: Obligatory \(t\)-SNE plot of PBMC dataset based on its ADT expression values, where each point is a cell and is colored by the cluster of origin. Cluster labels are also overlaid at the median coordinates across all cells in the cluster.
We perform some additional subclustering using the expression data to mimic an in silico FACS experiment.
set.seed(1010010)
subclusters <- quickSubCluster(sce.pbmc, clust.adt,
prepFUN=function(x) {
dec <- modelGeneVarByPoisson(x)
top <- getTopHVGs(dec, prop=0.1)
denoisePCA(x, dec, subset.row=top)
},
clusterFUN=function(x) {
g.gene <- buildSNNGraph(x, k=10, use.dimred = 'PCA')
igraph::cluster_walktrap(g.gene)$membership
}
)We counting the number of gene expression-derived subclusters in each ADT-derived parent cluster.
data.frame(
Cluster=names(subclusters),
Ncells=vapply(subclusters, ncol, 0L),
Nsub=vapply(subclusters, function(x) length(unique(x$subcluster)), 0L)
)## Cluster Ncells Nsub
## 1 1 160 3
## 2 2 507 4
## 3 3 662 4
## 4 4 39 1
## 5 5 691 5
## 6 6 1415 4
## 7 7 32 1
## 8 8 650 7
## 9 9 76 2
## 10 10 1037 9
## 11 11 121 2
## 12 12 47 1
## 13 13 68 2
## 14 14 25 1
## 15 15 15 1
## 16 16 562 6
## 17 17 139 3
## 18 18 32 1
## 19 19 44 1
## 20 20 120 4
## 21 21 84 2
## 22 22 65 2
## 23 23 52 2
## 24 24 17 1Session Info
R version 4.4.0 beta (2024-04-15 r86425)
Platform: x86_64-pc-linux-gnu
Running under: Ubuntu 22.04.4 LTS
Matrix products: default
BLAS: /home/biocbuild/bbs-3.19-bioc/R/lib/libRblas.so
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.10.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_GB LC_COLLATE=C
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
time zone: America/New_York
tzcode source: system (glibc)
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] pheatmap_1.0.12 bluster_1.14.0
[3] scran_1.32.0 scater_1.32.0
[5] ggplot2_3.5.1 scuttle_1.14.0
[7] DropletUtils_1.24.0 SingleCellExperiment_1.26.0
[9] SummarizedExperiment_1.34.0 Biobase_2.64.0
[11] GenomicRanges_1.56.0 GenomeInfoDb_1.40.0
[13] IRanges_2.38.0 S4Vectors_0.42.0
[15] BiocGenerics_0.50.0 MatrixGenerics_1.16.0
[17] matrixStats_1.3.0 BiocFileCache_2.12.0
[19] dbplyr_2.5.0 BiocStyle_2.32.0
[21] rebook_1.14.0
loaded via a namespace (and not attached):
[1] DBI_1.2.2 gridExtra_2.3
[3] CodeDepends_0.6.6 rlang_1.1.3
[5] magrittr_2.0.3 RcppAnnoy_0.0.22
[7] compiler_4.4.0 RSQLite_2.3.6
[9] dir.expiry_1.12.0 DelayedMatrixStats_1.26.0
[11] vctrs_0.6.5 pkgconfig_2.0.3
[13] crayon_1.5.2 fastmap_1.1.1
[15] XVector_0.44.0 labeling_0.4.3
[17] utf8_1.2.4 rmarkdown_2.26
[19] ggbeeswarm_0.7.2 graph_1.82.0
[21] UCSC.utils_1.0.0 purrr_1.0.2
[23] bit_4.0.5 xfun_0.43
[25] zlibbioc_1.50.0 cachem_1.0.8
[27] beachmat_2.20.0 jsonlite_1.8.8
[29] blob_1.2.4 highr_0.10
[31] rhdf5filters_1.16.0 DelayedArray_0.30.0
[33] Rhdf5lib_1.26.0 BiocParallel_1.38.0
[35] cluster_2.1.6 irlba_2.3.5.1
[37] parallel_4.4.0 R6_2.5.1
[39] RColorBrewer_1.1-3 bslib_0.7.0
[41] limma_3.60.0 jquerylib_0.1.4
[43] Rcpp_1.0.12 bookdown_0.39
[45] knitr_1.46 R.utils_2.12.3
[47] igraph_2.0.3 Matrix_1.7-0
[49] tidyselect_1.2.1 viridis_0.6.5
[51] abind_1.4-5 yaml_2.3.8
[53] codetools_0.2-20 curl_5.2.1
[55] lattice_0.22-6 tibble_3.2.1
[57] withr_3.0.0 Rtsne_0.17
[59] evaluate_0.23 pillar_1.9.0
[61] BiocManager_1.30.22 filelock_1.0.3
[63] generics_0.1.3 sparseMatrixStats_1.16.0
[65] munsell_0.5.1 scales_1.3.0
[67] glue_1.7.0 metapod_1.12.0
[69] tools_4.4.0 BiocNeighbors_1.22.0
[71] ScaledMatrix_1.12.0 locfit_1.5-9.9
[73] XML_3.99-0.16.1 cowplot_1.1.3
[75] rhdf5_2.48.0 grid_4.4.0
[77] edgeR_4.2.0 colorspace_2.1-0
[79] GenomeInfoDbData_1.2.12 beeswarm_0.4.0
[81] BiocSingular_1.20.0 HDF5Array_1.32.0
[83] vipor_0.4.7 cli_3.6.2
[85] rsvd_1.0.5 fansi_1.0.6
[87] viridisLite_0.4.2 S4Arrays_1.4.0
[89] dplyr_1.1.4 uwot_0.2.2
[91] gtable_0.3.5 R.methodsS3_1.8.2
[93] sass_0.4.9 digest_0.6.35
[95] ggrepel_0.9.5 SparseArray_1.4.0
[97] dqrng_0.3.2 farver_2.1.1
[99] memoise_2.0.1 htmltools_0.5.8.1
[101] R.oo_1.26.0 lifecycle_1.0.4
[103] httr_1.4.7 statmod_1.5.0
[105] bit64_4.0.5 References
Zheng, G. X., J. M. Terry, P. Belgrader, P. Ryvkin, Z. W. Bent, R. Wilson, S. B. Ziraldo, et al. 2017. “Massively parallel digital transcriptional profiling of single cells.” Nat Commun 8 (January): 14049.