Chapter 5 Analysis overview
5.1 Outline
This chapter provides an overview of the framework of a typical scRNA-seq analysis workflow (Figure 5.1).
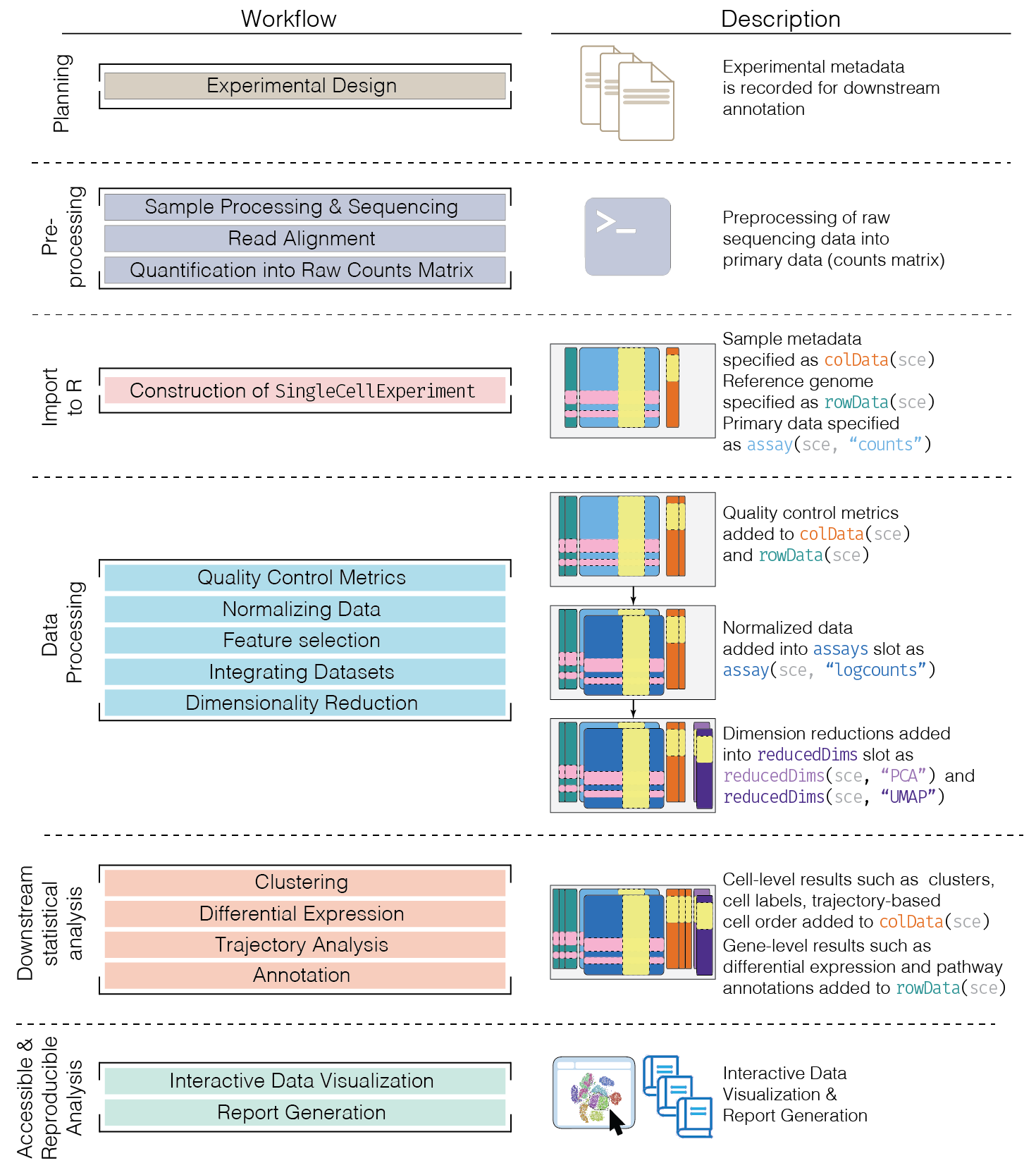
Figure 5.1: Schematic of a typical scRNA-seq analysis workflow. Each stage (separated by dashed lines) consists of a number of specific steps, many of which operate on and modify a SingleCellExperiment instance.
In the simplest case, the workflow has the following form:
- We compute quality control metrics to remove low-quality cells that would interfere with downstream analyses. These cells may have been damaged during processing or may not have been fully captured by the sequencing protocol. Common metrics includes the total counts per cell, the proportion of spike-in or mitochondrial reads and the number of detected features.
- We convert the counts into normalized expression values to eliminate cell-specific biases (e.g., in capture efficiency). This allows us to perform explicit comparisons across cells in downstream steps like clustering. We also apply a transformation, typically log, to adjust for the mean-variance relationship.
- We perform feature selection to pick a subset of interesting features for downstream analysis. This is done by modelling the variance across cells for each gene and retaining genes that are highly variable. The aim is to reduce computational overhead and noise from uninteresting genes.
- We apply dimensionality reduction to compact the data and further reduce noise. Principal components analysis is typically used to obtain an initial low-rank representation for more computational work, followed by more aggressive methods like \(t\)-stochastic neighbor embedding for visualization purposes.
- We cluster cells into groups according to similarities in their (normalized) expression profiles. This aims to obtain groupings that serve as empirical proxies for distinct biological states. We typically interpret these groupings by identifying differentially expressed marker genes between clusters.
Subsequent chapters will describe each analysis step in more detail.
5.2 Quick start (simple)
Here, we use the a droplet-based retina dataset from Macosko et al. (2015), provided in the scRNAseq package. This starts from a count matrix and finishes with clusters (Figure 5.2) in preparation for biological interpretation. Similar workflows are available in abbreviated form in later parts of the book.
library(scRNAseq)
sce <- MacoskoRetinaData()
# Quality control (using mitochondrial genes).
library(scater)
is.mito <- grepl("^MT-", rownames(sce))
qcstats <- perCellQCMetrics(sce, subsets=list(Mito=is.mito))
filtered <- quickPerCellQC(qcstats, percent_subsets="subsets_Mito_percent")
sce <- sce[, !filtered$discard]
# Normalization.
sce <- logNormCounts(sce)
# Feature selection.
library(scran)
dec <- modelGeneVar(sce)
hvg <- getTopHVGs(dec, prop=0.1)
# PCA.
library(scater)
set.seed(1234)
sce <- runPCA(sce, ncomponents=25, subset_row=hvg)
# Clustering.
library(bluster)
colLabels(sce) <- clusterCells(sce, use.dimred='PCA',
BLUSPARAM=NNGraphParam(cluster.fun="louvain"))
# Visualization.
sce <- runUMAP(sce, dimred = 'PCA')
plotUMAP(sce, colour_by="label")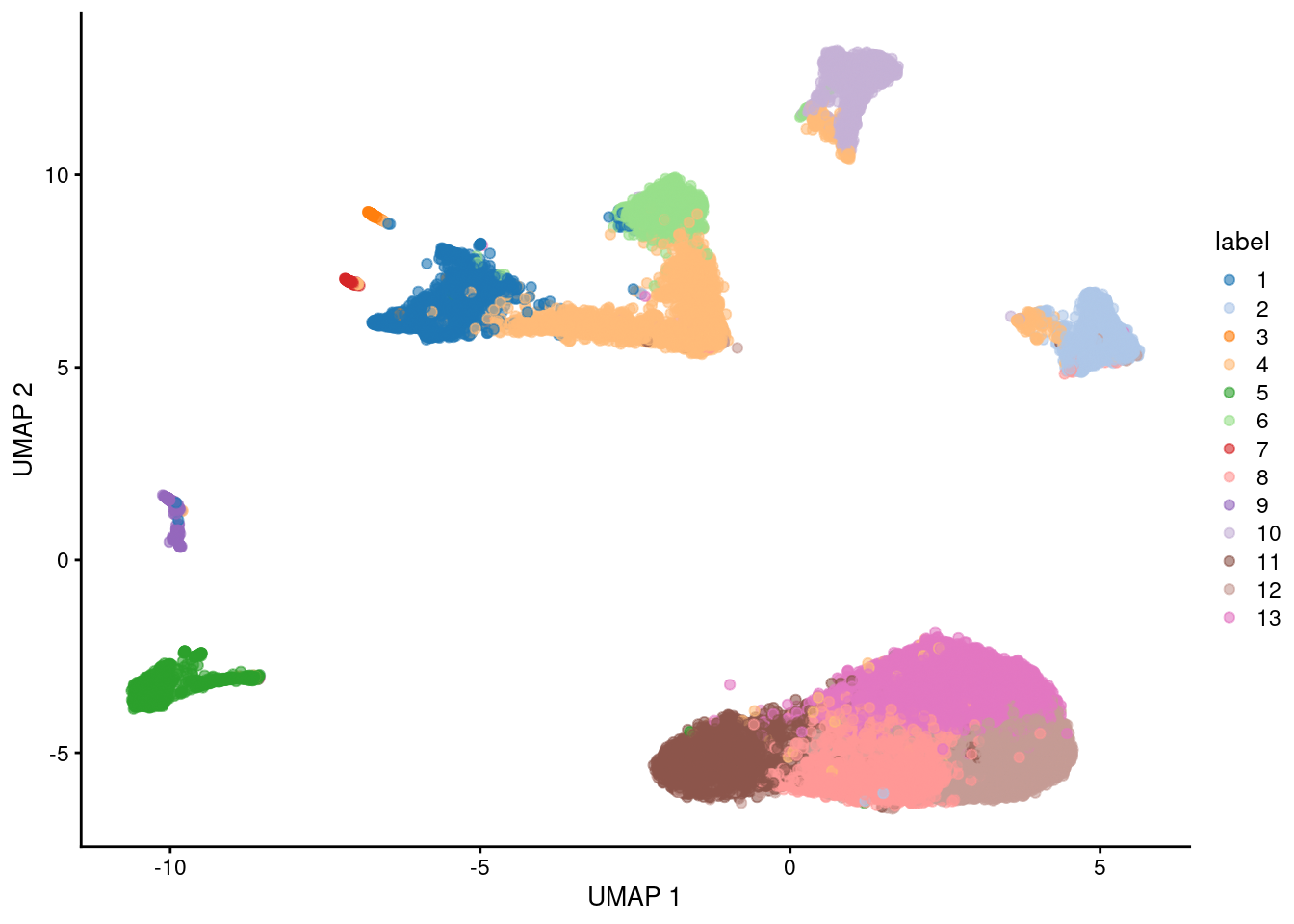
Figure 5.2: UMAP plot of the retina dataset, where each point is a cell and is colored by the assigned cluster identity.
5.3 Quick start (multiple batches)
Here we use the pancreas Smart-seq2 dataset from Segerstolpe et al. (2016), again provided in the scRNAseq package. This starts from a count matrix and finishes with clusters (Figure 5.2) with some additional tweaks to eliminate uninteresting batch effects between individuals. Note that a more elaborate analysis of the same dataset with justifications for each step is available in Workflow Chapter 8.
sce <- SegerstolpePancreasData()
# Quality control (using ERCCs).
qcstats <- perCellQCMetrics(sce)
filtered <- quickPerCellQC(qcstats, percent_subsets="altexps_ERCC_percent")
sce <- sce[, !filtered$discard]
# Normalization.
sce <- logNormCounts(sce)
# Feature selection, blocking on the individual of origin.
dec <- modelGeneVar(sce, block=sce$individual)
hvg <- getTopHVGs(dec, prop=0.1)
# Batch correction.
library(batchelor)
set.seed(1234)
sce <- correctExperiments(sce, batch=sce$individual,
subset.row=hvg, correct.all=TRUE)
# Clustering.
colLabels(sce) <- clusterCells(sce, use.dimred='corrected')
# Visualization.
sce <- runUMAP(sce, dimred = 'corrected')
gridExtra::grid.arrange(
plotUMAP(sce, colour_by="label"),
plotUMAP(sce, colour_by="individual"),
ncol=2
)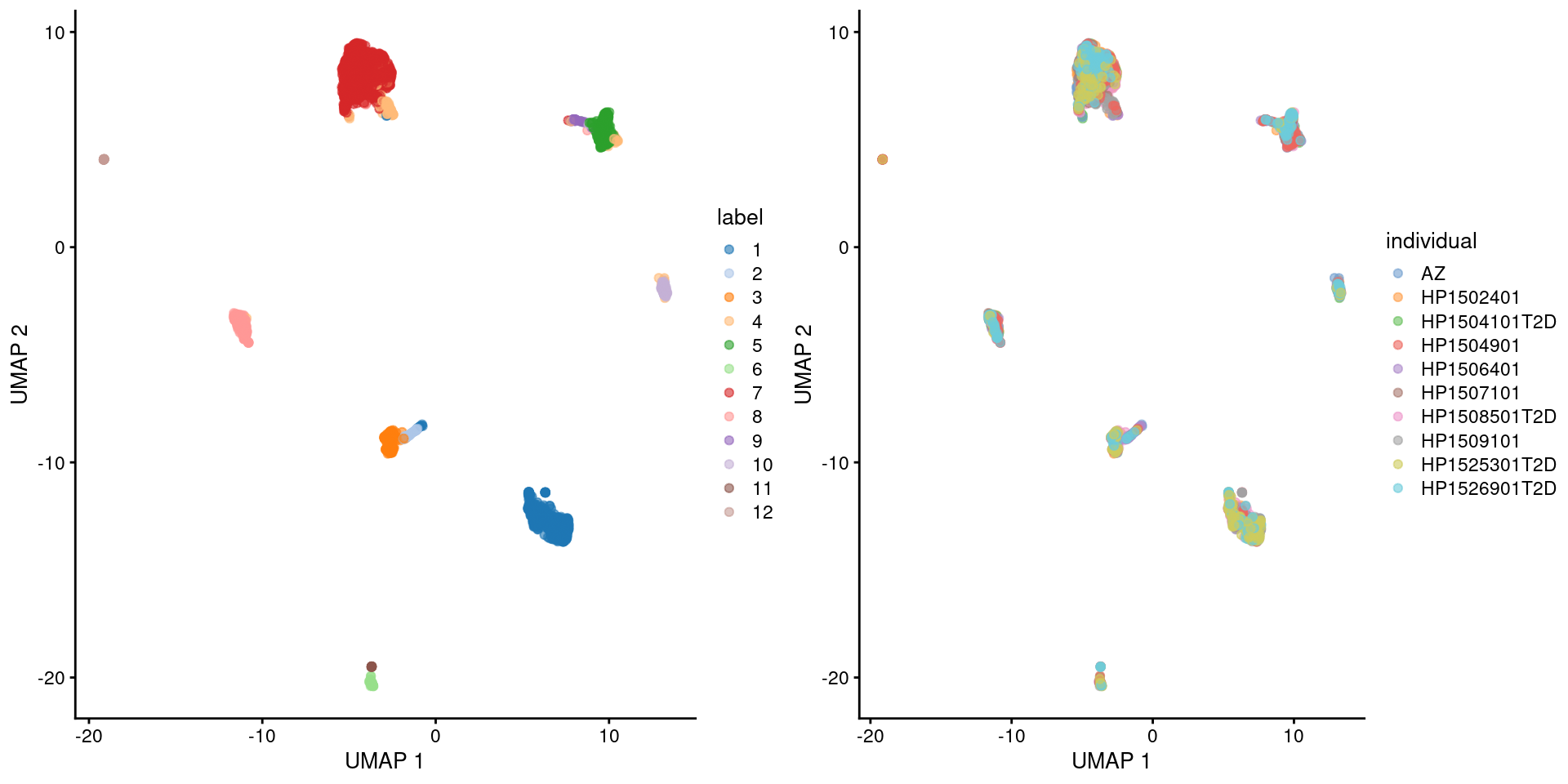
Figure 5.3: UMAP plot of the pancreas dataset, where each point is a cell and is colored by the assigned cluster identity (left) or the individual of origin (right).
Session Info
R version 4.3.1 (2023-06-16)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 22.04.3 LTS
Matrix products: default
BLAS: /home/biocbuild/bbs-3.18-bioc/R/lib/libRblas.so
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.10.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_GB LC_COLLATE=C
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
time zone: America/New_York
tzcode source: system (glibc)
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] BiocSingular_1.18.0 batchelor_1.18.0
[3] bluster_1.12.0 scran_1.30.0
[5] scater_1.30.0 ggplot2_3.4.4
[7] scuttle_1.12.0 Matrix_1.6-1.1
[9] scRNAseq_2.15.0 SingleCellExperiment_1.24.0
[11] SummarizedExperiment_1.32.0 Biobase_2.62.0
[13] GenomicRanges_1.54.0 GenomeInfoDb_1.38.0
[15] IRanges_2.36.0 S4Vectors_0.40.0
[17] BiocGenerics_0.48.0 MatrixGenerics_1.14.0
[19] matrixStats_1.0.0 BiocStyle_2.30.0
[21] rebook_1.12.0
loaded via a namespace (and not attached):
[1] rstudioapi_0.15.0 jsonlite_1.8.7
[3] CodeDepends_0.6.5 magrittr_2.0.3
[5] ggbeeswarm_0.7.2 GenomicFeatures_1.54.0
[7] farver_2.1.1 rmarkdown_2.25
[9] BiocIO_1.12.0 zlibbioc_1.48.0
[11] vctrs_0.6.4 memoise_2.0.1
[13] Rsamtools_2.18.0 DelayedMatrixStats_1.24.0
[15] RCurl_1.98-1.12 htmltools_0.5.6.1
[17] S4Arrays_1.2.0 progress_1.2.2
[19] AnnotationHub_3.10.0 curl_5.1.0
[21] BiocNeighbors_1.20.0 SparseArray_1.2.0
[23] sass_0.4.7 bslib_0.5.1
[25] cachem_1.0.8 ResidualMatrix_1.12.0
[27] GenomicAlignments_1.38.0 igraph_1.5.1
[29] mime_0.12 lifecycle_1.0.3
[31] pkgconfig_2.0.3 rsvd_1.0.5
[33] R6_2.5.1 fastmap_1.1.1
[35] GenomeInfoDbData_1.2.11 shiny_1.7.5.1
[37] digest_0.6.33 colorspace_2.1-0
[39] AnnotationDbi_1.64.0 dqrng_0.3.1
[41] irlba_2.3.5.1 ExperimentHub_2.10.0
[43] RSQLite_2.3.1 beachmat_2.18.0
[45] labeling_0.4.3 filelock_1.0.2
[47] fansi_1.0.5 httr_1.4.7
[49] abind_1.4-5 compiler_4.3.1
[51] bit64_4.0.5 withr_2.5.1
[53] BiocParallel_1.36.0 viridis_0.6.4
[55] DBI_1.1.3 biomaRt_2.58.0
[57] rappdirs_0.3.3 DelayedArray_0.28.0
[59] rjson_0.2.21 tools_4.3.1
[61] vipor_0.4.5 beeswarm_0.4.0
[63] interactiveDisplayBase_1.40.0 httpuv_1.6.12
[65] glue_1.6.2 restfulr_0.0.15
[67] promises_1.2.1 grid_4.3.1
[69] cluster_2.1.4 generics_0.1.3
[71] gtable_0.3.4 ensembldb_2.26.0
[73] hms_1.1.3 metapod_1.10.0
[75] ScaledMatrix_1.10.0 xml2_1.3.5
[77] utf8_1.2.4 XVector_0.42.0
[79] RcppAnnoy_0.0.21 ggrepel_0.9.4
[81] BiocVersion_3.18.0 pillar_1.9.0
[83] stringr_1.5.0 limma_3.58.0
[85] later_1.3.1 dplyr_1.1.3
[87] BiocFileCache_2.10.0 lattice_0.22-5
[89] FNN_1.1.3.2 rtracklayer_1.62.0
[91] bit_4.0.5 tidyselect_1.2.0
[93] locfit_1.5-9.8 Biostrings_2.70.0
[95] knitr_1.44 gridExtra_2.3
[97] bookdown_0.36 ProtGenerics_1.34.0
[99] edgeR_4.0.0 xfun_0.40
[101] statmod_1.5.0 stringi_1.7.12
[103] lazyeval_0.2.2 yaml_2.3.7
[105] evaluate_0.22 codetools_0.2-19
[107] tibble_3.2.1 BiocManager_1.30.22
[109] graph_1.80.0 cli_3.6.1
[111] uwot_0.1.16 xtable_1.8-4
[113] munsell_0.5.0 jquerylib_0.1.4
[115] Rcpp_1.0.11 dir.expiry_1.10.0
[117] dbplyr_2.3.4 png_0.1-8
[119] XML_3.99-0.14 parallel_4.3.1
[121] ellipsis_0.3.2 blob_1.2.4
[123] prettyunits_1.2.0 AnnotationFilter_1.26.0
[125] sparseMatrixStats_1.14.0 bitops_1.0-7
[127] viridisLite_0.4.2 scales_1.2.1
[129] purrr_1.0.2 crayon_1.5.2
[131] rlang_1.1.1 cowplot_1.1.1
[133] KEGGREST_1.42.0 Islam, S., A. Zeisel, S. Joost, G. La Manno, P. Zajac, M. Kasper, P. Lonnerberg, and S. Linnarsson. 2014. “Quantitative single-cell RNA-seq with unique molecular identifiers.” Nat. Methods 11 (2): 163–66.
Lun, A. T. L., F. J. Calero-Nieto, L. Haim-Vilmovsky, B. Gottgens, and J. C. Marioni. 2017. “Assessing the reliability of spike-in normalization for analyses of single-cell RNA sequencing data.” Genome Res. 27 (11): 1795–1806.
Macosko, E. Z., A. Basu, R. Satija, J. Nemesh, K. Shekhar, M. Goldman, I. Tirosh, et al. 2015. “Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets.” Cell 161 (5): 1202–14.
Mereu, Elisabetta, Atefeh Lafzi, Catia Moutinho, Christoph Ziegenhain, Davis J. MacCarthy, Adrian Alvarez, Eduard Batlle, et al. 2019. “Benchmarking Single-Cell Rna Sequencing Protocols for Cell Atlas Projects.” bioRxiv. https://doi.org/10.1101/630087.
Muraro, M. J., G. Dharmadhikari, D. Grun, N. Groen, T. Dielen, E. Jansen, L. van Gurp, et al. 2016. “A Single-Cell Transcriptome Atlas of the Human Pancreas.” Cell Syst 3 (4): 385–94.
Segerstolpe, A., A. Palasantza, P. Eliasson, E. M. Andersson, A. C. Andreasson, X. Sun, S. Picelli, et al. 2016. “Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes.” Cell Metab. 24 (4): 593–607.
Srivastava, A., L. Malik, T. Smith, I. Sudbery, and R. Patro. 2019. “Alevin efficiently estimates accurate gene abundances from dscRNA-seq data.” Genome Biol 20 (1): 65.
Svensson, V., E. da Veiga Beltrame, and L. Pachter. 2019. “Quantifying the Tradeoff Between Sequencing Depth and Cell Number in Single-Cell Rna-Seq.” bioRxiv, 762773.
Wilson, N. K., D. G. Kent, F. Buettner, M. Shehata, I. C. Macaulay, F. J. Calero-Nieto, M. Sanchez Castillo, et al. 2015. “Combined single-cell functional and gene expression analysis resolves heterogeneity within stem cell populations.” Cell Stem Cell 16 (6): 712–24.
Zhang, M. J., V. Ntranos, and D. Tse. 2020. “Determining sequencing depth in a single-cell RNA-seq experiment.” Nat Commun 11 (1): 774.
Zheng, G. X., J. M. Terry, P. Belgrader, P. Ryvkin, Z. W. Bent, R. Wilson, S. B. Ziraldo, et al. 2017. “Massively parallel digital transcriptional profiling of single cells.” Nat Commun 8 (January): 14049.
Ziegenhain, C., B. Vieth, S. Parekh, B. Reinius, A. Guillaumet-Adkins, M. Smets, H. Leonhardt, H. Heyn, I. Hellmann, and W. Enard. 2017. “Comparative Analysis of Single-Cell RNA Sequencing Methods.” Mol. Cell 65 (4): 631–43.
References
Macosko, E. Z., A. Basu, R. Satija, J. Nemesh, K. Shekhar, M. Goldman, I. Tirosh, et al. 2015. “Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets.” Cell 161 (5): 1202–14.
Segerstolpe, A., A. Palasantza, P. Eliasson, E. M. Andersson, A. C. Andreasson, X. Sun, S. Picelli, et al. 2016. “Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes.” Cell Metab. 24 (4): 593–607.