Chapter 6 Muraro human pancreas (CEL-seq)
6.1 Introduction
This performs an analysis of the Muraro et al. (2016) CEL-seq dataset, consisting of human pancreas cells from various donors.
6.2 Data loading
Converting back to Ensembl identifiers.
library(AnnotationHub)
edb <- AnnotationHub()[["AH73881"]]
gene.symb <- sub("__chr.*$", "", rownames(sce.muraro))
gene.ids <- mapIds(edb, keys=gene.symb,
keytype="SYMBOL", column="GENEID")
# Removing duplicated genes or genes without Ensembl IDs.
keep <- !is.na(gene.ids) & !duplicated(gene.ids)
sce.muraro <- sce.muraro[keep,]
rownames(sce.muraro) <- gene.ids[keep]6.3 Quality control
This dataset lacks mitochondrial genes so we will do without. For the one batch that seems to have a high proportion of low-quality cells, we compute an appropriate filter threshold using a shared median and MAD from the other batches (Figure 6.1).
library(scater)
stats <- perCellQCMetrics(sce.muraro)
qc <- quickPerCellQC(stats, percent_subsets="altexps_ERCC_percent",
batch=sce.muraro$donor, subset=sce.muraro$donor!="D28")
sce.muraro <- sce.muraro[,!qc$discard]colData(unfiltered) <- cbind(colData(unfiltered), stats)
unfiltered$discard <- qc$discard
gridExtra::grid.arrange(
plotColData(unfiltered, x="donor", y="sum", colour_by="discard") +
scale_y_log10() + ggtitle("Total count"),
plotColData(unfiltered, x="donor", y="detected", colour_by="discard") +
scale_y_log10() + ggtitle("Detected features"),
plotColData(unfiltered, x="donor", y="altexps_ERCC_percent",
colour_by="discard") + ggtitle("ERCC percent"),
ncol=2
)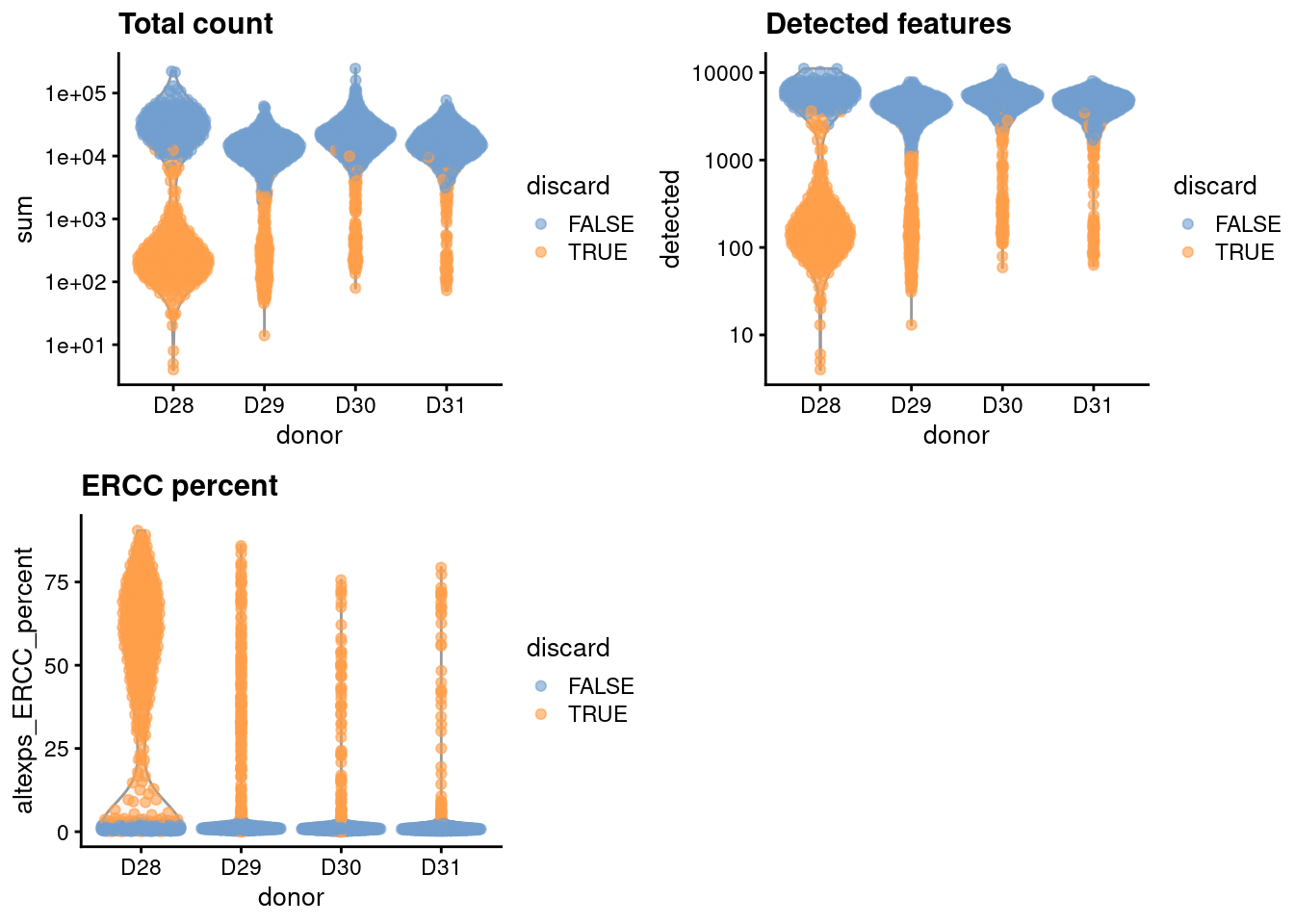
Figure 6.1: Distribution of each QC metric across cells from each donor in the Muraro pancreas dataset. Each point represents a cell and is colored according to whether that cell was discarded.
We have a look at the causes of removal:
## low_lib_size low_n_features high_altexps_ERCC_percent
## 663 700 738
## discard
## 7736.4 Normalization
library(scran)
set.seed(1000)
clusters <- quickCluster(sce.muraro)
sce.muraro <- computeSumFactors(sce.muraro, clusters=clusters)
sce.muraro <- logNormCounts(sce.muraro)## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.088 0.541 0.821 1.000 1.211 13.987plot(librarySizeFactors(sce.muraro), sizeFactors(sce.muraro), pch=16,
xlab="Library size factors", ylab="Deconvolution factors", log="xy")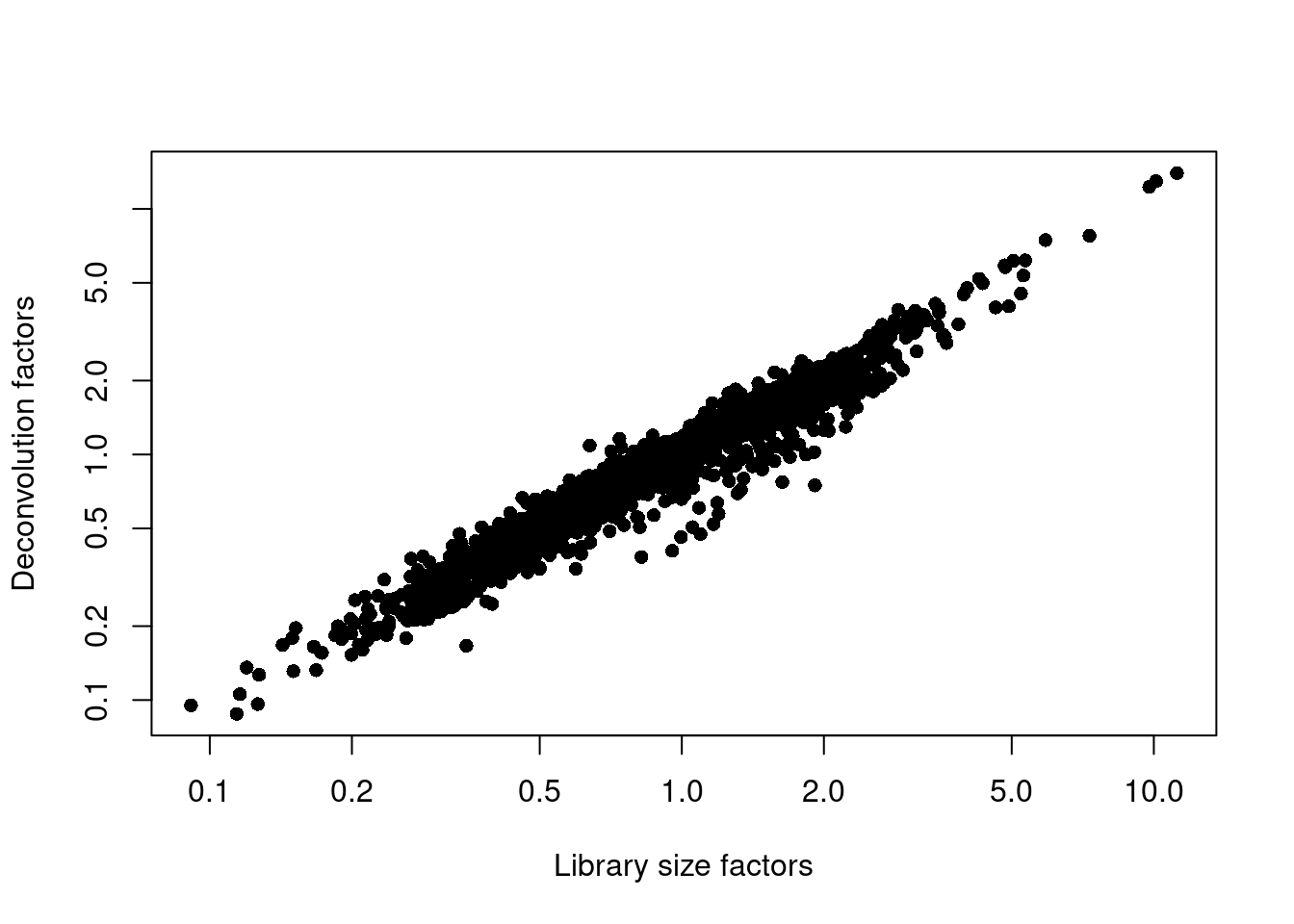
Figure 6.2: Relationship between the library size factors and the deconvolution size factors in the Muraro pancreas dataset.
6.5 Variance modelling
We block on a combined plate and donor factor.
block <- paste0(sce.muraro$plate, "_", sce.muraro$donor)
dec.muraro <- modelGeneVarWithSpikes(sce.muraro, "ERCC", block=block)
top.muraro <- getTopHVGs(dec.muraro, prop=0.1)par(mfrow=c(8,4))
blocked.stats <- dec.muraro$per.block
for (i in colnames(blocked.stats)) {
current <- blocked.stats[[i]]
plot(current$mean, current$total, main=i, pch=16, cex=0.5,
xlab="Mean of log-expression", ylab="Variance of log-expression")
curfit <- metadata(current)
points(curfit$mean, curfit$var, col="red", pch=16)
curve(curfit$trend(x), col='dodgerblue', add=TRUE, lwd=2)
}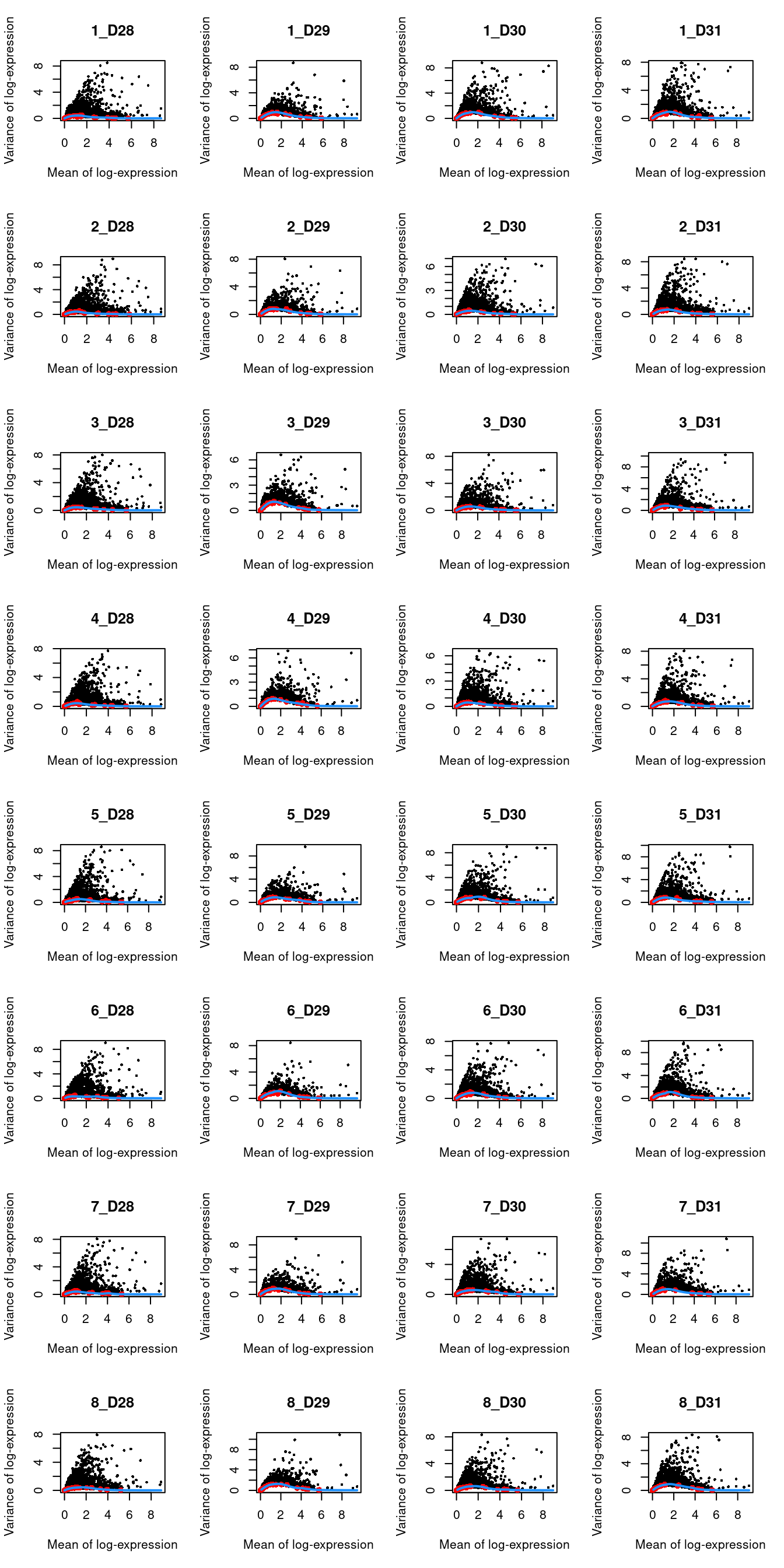
Figure 6.3: Per-gene variance as a function of the mean for the log-expression values in the Muraro pancreas dataset. Each point represents a gene (black) with the mean-variance trend (blue) fitted to the spike-in transcripts (red) separately for each donor.
6.6 Data integration
library(batchelor)
set.seed(1001010)
merged.muraro <- fastMNN(sce.muraro, subset.row=top.muraro,
batch=sce.muraro$donor)We use the proportion of variance lost as a diagnostic measure:
## D28 D29 D30 D31
## [1,] 0.060847 0.024121 0.000000 0.00000
## [2,] 0.002646 0.003018 0.062421 0.00000
## [3,] 0.003449 0.002641 0.002598 0.081626.7 Dimensionality reduction
6.8 Clustering
snn.gr <- buildSNNGraph(merged.muraro, use.dimred="corrected")
colLabels(merged.muraro) <- factor(igraph::cluster_walktrap(snn.gr)$membership)tab <- table(Cluster=colLabels(merged.muraro), CellType=sce.muraro$label)
library(pheatmap)
pheatmap(log10(tab+10), color=viridis::viridis(100))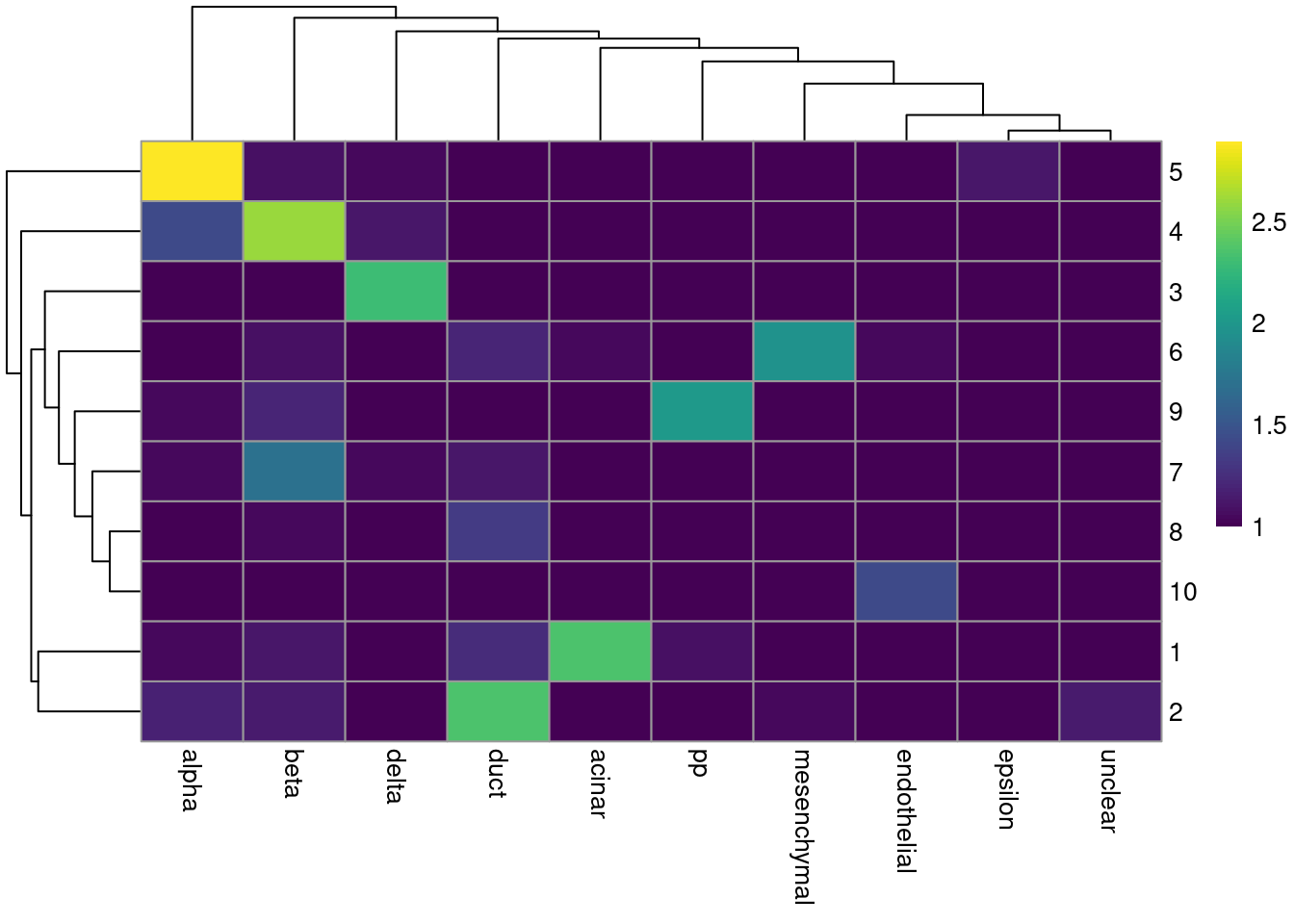
Figure 6.4: Heatmap of the frequency of cells from each cell type label in each cluster.
## Donor
## Cluster D28 D29 D30 D31
## 1 104 6 57 112
## 2 59 21 77 97
## 3 12 75 64 43
## 4 28 149 126 120
## 5 87 261 277 214
## 6 21 7 54 26
## 7 1 6 6 37
## 8 6 6 5 2
## 9 11 68 5 30
## 10 4 2 5 8gridExtra::grid.arrange(
plotTSNE(merged.muraro, colour_by="label"),
plotTSNE(merged.muraro, colour_by="batch"),
ncol=2
)
Figure 6.5: Obligatory \(t\)-SNE plots of the Muraro pancreas dataset. Each point represents a cell that is colored by cluster (left) or batch (right).
Session Info
R version 4.1.0 (2021-05-18)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 20.04.2 LTS
Matrix products: default
BLAS: /home/biocbuild/bbs-3.13-bioc/R/lib/libRblas.so
LAPACK: /home/biocbuild/bbs-3.13-bioc/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=C
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] pheatmap_1.0.12 batchelor_1.8.0
[3] scran_1.20.0 scater_1.20.0
[5] ggplot2_3.3.3 scuttle_1.2.0
[7] ensembldb_2.16.0 AnnotationFilter_1.16.0
[9] GenomicFeatures_1.44.0 AnnotationDbi_1.54.0
[11] AnnotationHub_3.0.0 BiocFileCache_2.0.0
[13] dbplyr_2.1.1 scRNAseq_2.6.0
[15] SingleCellExperiment_1.14.0 SummarizedExperiment_1.22.0
[17] Biobase_2.52.0 GenomicRanges_1.44.0
[19] GenomeInfoDb_1.28.0 IRanges_2.26.0
[21] S4Vectors_0.30.0 BiocGenerics_0.38.0
[23] MatrixGenerics_1.4.0 matrixStats_0.58.0
[25] BiocStyle_2.20.0 rebook_1.2.0
loaded via a namespace (and not attached):
[1] igraph_1.2.6 lazyeval_0.2.2
[3] BiocParallel_1.26.0 digest_0.6.27
[5] htmltools_0.5.1.1 viridis_0.6.1
[7] fansi_0.4.2 magrittr_2.0.1
[9] memoise_2.0.0 ScaledMatrix_1.0.0
[11] cluster_2.1.2 limma_3.48.0
[13] Biostrings_2.60.0 prettyunits_1.1.1
[15] colorspace_2.0-1 blob_1.2.1
[17] rappdirs_0.3.3 xfun_0.23
[19] dplyr_1.0.6 crayon_1.4.1
[21] RCurl_1.98-1.3 jsonlite_1.7.2
[23] graph_1.70.0 glue_1.4.2
[25] gtable_0.3.0 zlibbioc_1.38.0
[27] XVector_0.32.0 DelayedArray_0.18.0
[29] BiocSingular_1.8.0 scales_1.1.1
[31] edgeR_3.34.0 DBI_1.1.1
[33] Rcpp_1.0.6 viridisLite_0.4.0
[35] xtable_1.8-4 progress_1.2.2
[37] dqrng_0.3.0 bit_4.0.4
[39] rsvd_1.0.5 ResidualMatrix_1.2.0
[41] metapod_1.0.0 httr_1.4.2
[43] RColorBrewer_1.1-2 dir.expiry_1.0.0
[45] ellipsis_0.3.2 pkgconfig_2.0.3
[47] XML_3.99-0.6 farver_2.1.0
[49] CodeDepends_0.6.5 sass_0.4.0
[51] locfit_1.5-9.4 utf8_1.2.1
[53] tidyselect_1.1.1 labeling_0.4.2
[55] rlang_0.4.11 later_1.2.0
[57] munsell_0.5.0 BiocVersion_3.13.1
[59] tools_4.1.0 cachem_1.0.5
[61] generics_0.1.0 RSQLite_2.2.7
[63] ExperimentHub_2.0.0 evaluate_0.14
[65] stringr_1.4.0 fastmap_1.1.0
[67] yaml_2.2.1 knitr_1.33
[69] bit64_4.0.5 purrr_0.3.4
[71] KEGGREST_1.32.0 sparseMatrixStats_1.4.0
[73] mime_0.10 biomaRt_2.48.0
[75] compiler_4.1.0 beeswarm_0.3.1
[77] filelock_1.0.2 curl_4.3.1
[79] png_0.1-7 interactiveDisplayBase_1.30.0
[81] statmod_1.4.36 tibble_3.1.2
[83] bslib_0.2.5.1 stringi_1.6.2
[85] highr_0.9 bluster_1.2.0
[87] lattice_0.20-44 ProtGenerics_1.24.0
[89] Matrix_1.3-3 vctrs_0.3.8
[91] pillar_1.6.1 lifecycle_1.0.0
[93] BiocManager_1.30.15 jquerylib_0.1.4
[95] BiocNeighbors_1.10.0 cowplot_1.1.1
[97] bitops_1.0-7 irlba_2.3.3
[99] httpuv_1.6.1 rtracklayer_1.52.0
[101] R6_2.5.0 BiocIO_1.2.0
[103] bookdown_0.22 promises_1.2.0.1
[105] gridExtra_2.3 vipor_0.4.5
[107] codetools_0.2-18 assertthat_0.2.1
[109] rjson_0.2.20 withr_2.4.2
[111] GenomicAlignments_1.28.0 Rsamtools_2.8.0
[113] GenomeInfoDbData_1.2.6 hms_1.1.0
[115] grid_4.1.0 beachmat_2.8.0
[117] rmarkdown_2.8 DelayedMatrixStats_1.14.0
[119] Rtsne_0.15 shiny_1.6.0
[121] ggbeeswarm_0.6.0 restfulr_0.0.13 References
Muraro, M. J., G. Dharmadhikari, D. Grun, N. Groen, T. Dielen, E. Jansen, L. van Gurp, et al. 2016. “A Single-Cell Transcriptome Atlas of the Human Pancreas.” Cell Syst 3 (4): 385–94.