# Chimeric mouse embryo (10X Genomics)
## Introduction
This performs an analysis of the @pijuansala2019single dataset on mouse gastrulation.
Here, we examine chimeric embryos at the E8.5 stage of development
where td-Tomato-positive embryonic stem cells (ESCs) were injected into a wild-type blastocyst.
## Data loading
``` r
library(MouseGastrulationData)
sce.chimera <- WTChimeraData(samples=5:10)
sce.chimera
```
```
## class: SingleCellExperiment
## dim: 29453 20935
## metadata(0):
## assays(1): counts
## rownames(29453): ENSMUSG00000051951 ENSMUSG00000089699 ...
## ENSMUSG00000095742 tomato-td
## rowData names(2): ENSEMBL SYMBOL
## colnames(20935): cell_9769 cell_9770 ... cell_30702 cell_30703
## colData names(11): cell barcode ... doub.density sizeFactor
## reducedDimNames(2): pca.corrected.E7.5 pca.corrected.E8.5
## mainExpName: NULL
## altExpNames(0):
```
``` r
library(scater)
rownames(sce.chimera) <- uniquifyFeatureNames(
rowData(sce.chimera)$ENSEMBL, rowData(sce.chimera)$SYMBOL)
```
## Quality control
Quality control on the cells has already been performed by the authors, so we will not repeat it here.
We additionally remove cells that are labelled as stripped nuclei or doublets.
``` r
drop <- sce.chimera$celltype.mapped %in% c("stripped", "Doublet")
sce.chimera <- sce.chimera[,!drop]
```
## Normalization
We use the pre-computed size factors in `sce.chimera`.
``` r
sce.chimera <- logNormCounts(sce.chimera)
```
## Variance modelling
We retain all genes with any positive biological component, to preserve as much signal as possible across a very heterogeneous dataset.
``` r
library(scran)
dec.chimera <- modelGeneVar(sce.chimera, block=sce.chimera$sample)
chosen.hvgs <- dec.chimera$bio > 0
```
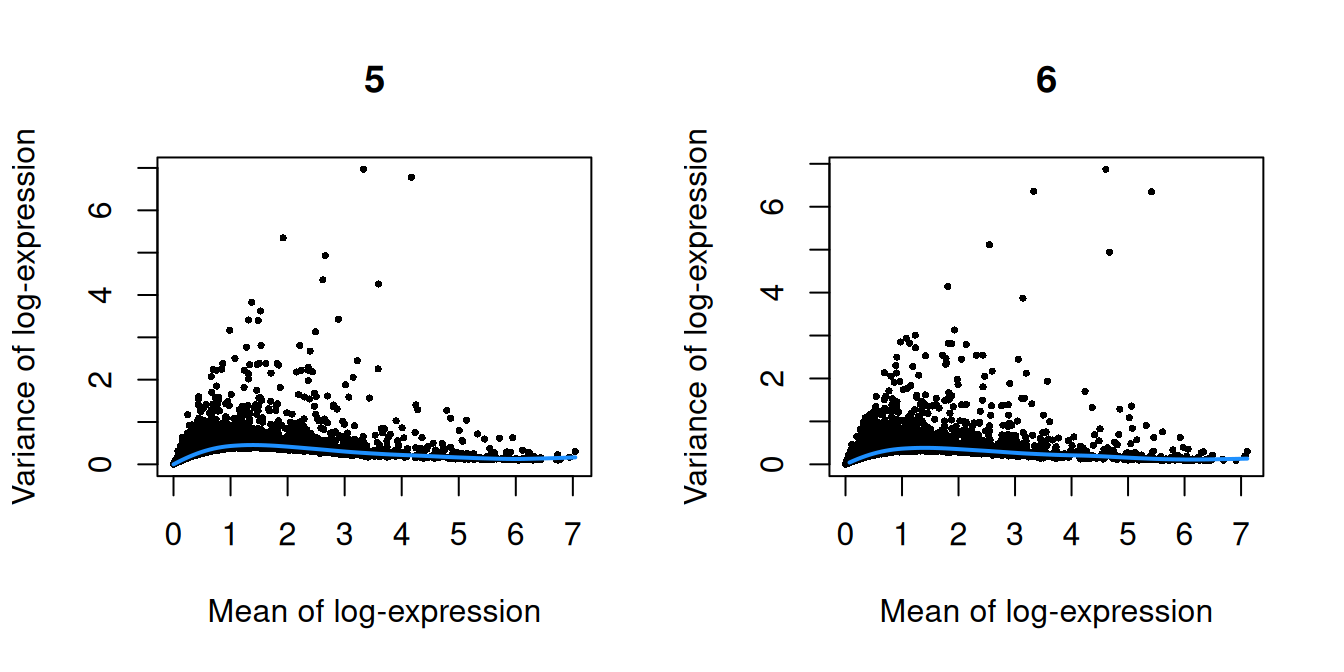
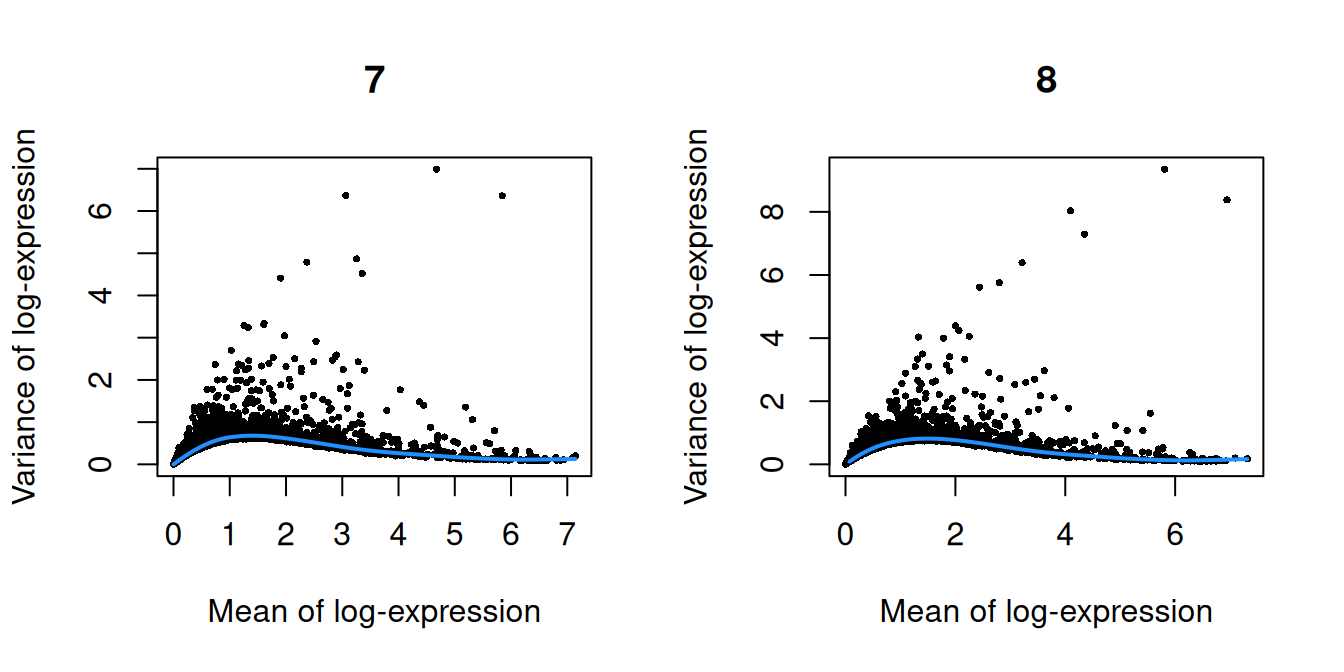
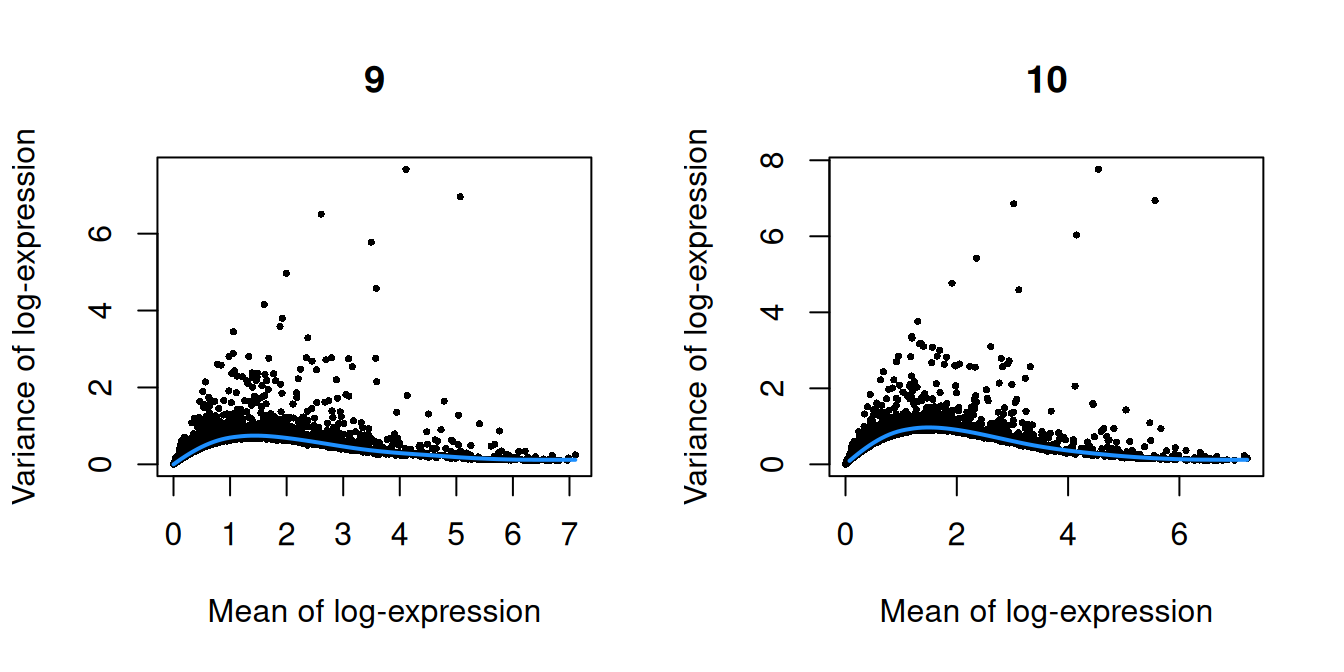
``` r
par(mfrow=c(1,2))
blocked.stats <- dec.chimera$per.block
for (i in colnames(blocked.stats)) {
current <- blocked.stats[[i]]
plot(current$mean, current$total, main=i, pch=16, cex=0.5,
xlab="Mean of log-expression", ylab="Variance of log-expression")
curfit <- metadata(current)
curve(curfit$trend(x), col='dodgerblue', add=TRUE, lwd=2)
}
```
(\#fig:unref-pijuan-var-1)Per-gene variance as a function of the mean for the log-expression values in the Pijuan-Sala chimeric mouse embryo dataset. Each point represents a gene (black) with the mean-variance trend (blue) fitted to the variances.
(\#fig:unref-pijuan-var-2)Per-gene variance as a function of the mean for the log-expression values in the Pijuan-Sala chimeric mouse embryo dataset. Each point represents a gene (black) with the mean-variance trend (blue) fitted to the variances.
(\#fig:unref-pijuan-var-3)Per-gene variance as a function of the mean for the log-expression values in the Pijuan-Sala chimeric mouse embryo dataset. Each point represents a gene (black) with the mean-variance trend (blue) fitted to the variances.
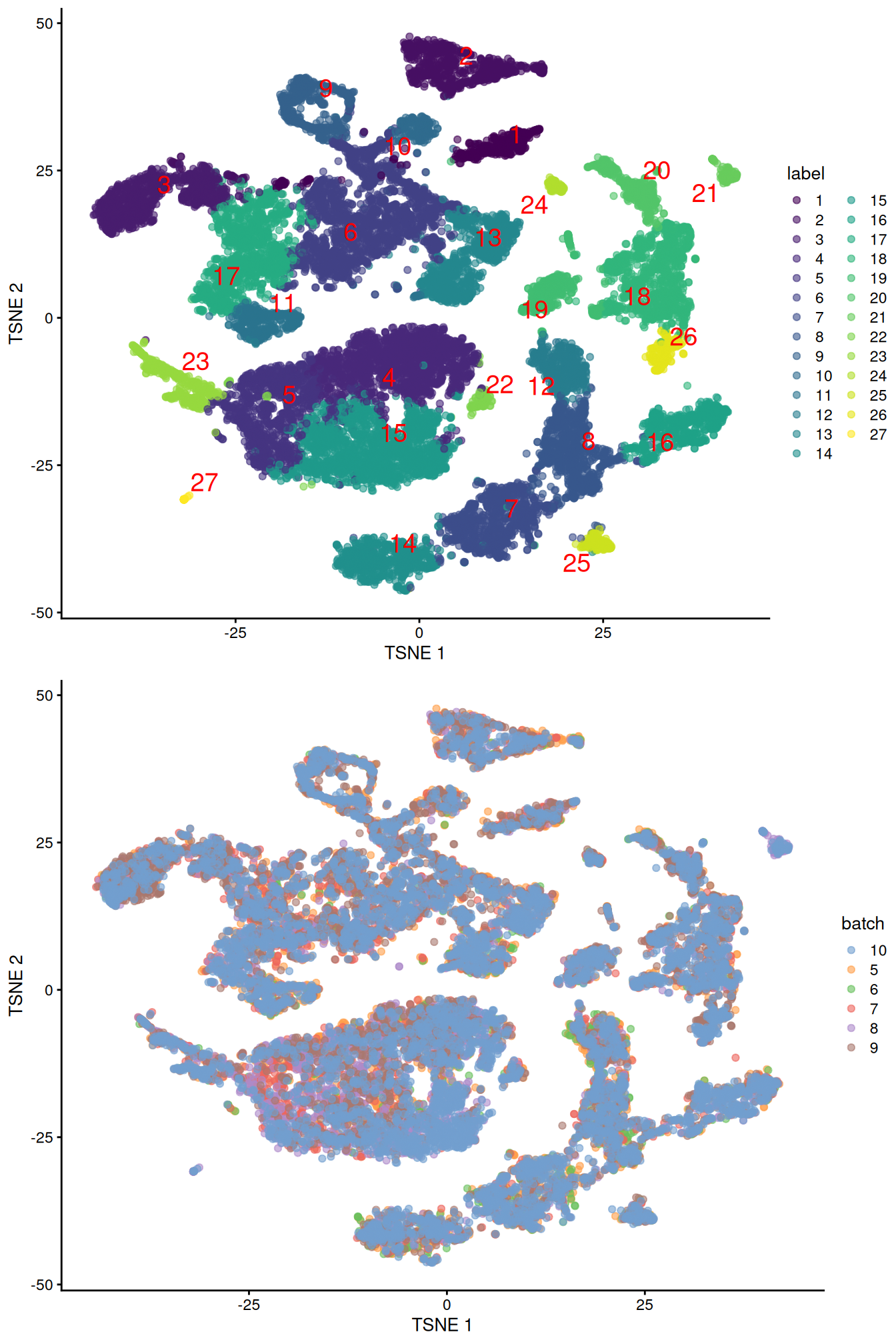
(\#fig:unref-pijuan-tsne)Obligatory $t$-SNE plots of the Pijuan-Sala chimeric mouse embryo dataset, where each point represents a cell and is colored according to the assigned cluster (top) or sample of origin (bottom).