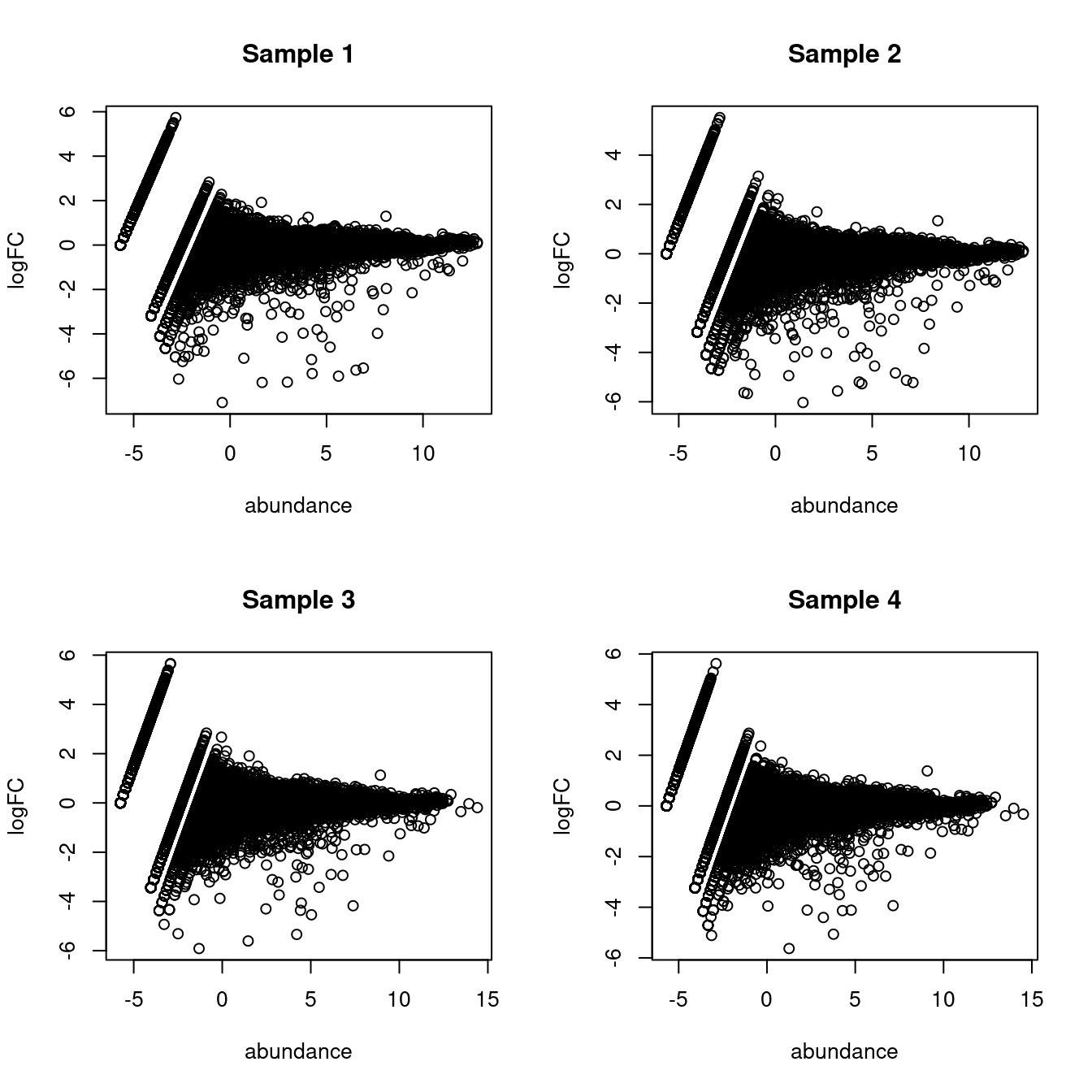
(\#fig:proxy-ambience)MA plots of the log-fold change of the proxy ambient profile over the real profile for each sample in the _Tal1_ chimera dataset.
```
R version 4.4.0 beta (2024-04-15 r86425)
Platform: x86_64-pc-linux-gnu
Running under: Ubuntu 22.04.4 LTS
Matrix products: default
BLAS: /home/biocbuild/bbs-3.19-bioc/R/lib/libRblas.so
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.10.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_GB LC_COLLATE=C
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
time zone: America/New_York
tzcode source: system (glibc)
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] edgeR_4.2.0 limma_3.60.0
[3] DropletUtils_1.24.0 scran_1.32.0
[5] scuttle_1.14.0 MouseGastrulationData_1.17.1
[7] SpatialExperiment_1.14.0 SingleCellExperiment_1.26.0
[9] SummarizedExperiment_1.34.0 Biobase_2.64.0
[11] GenomicRanges_1.56.0 GenomeInfoDb_1.40.0
[13] IRanges_2.38.0 S4Vectors_0.42.0
[15] BiocGenerics_0.50.0 MatrixGenerics_1.16.0
[17] matrixStats_1.3.0 BiocStyle_2.32.0
[19] rebook_1.14.0
loaded via a namespace (and not attached):
[1] DBI_1.2.2 CodeDepends_0.6.6
[3] rlang_1.1.3 magrittr_2.0.3
[5] compiler_4.4.0 RSQLite_2.3.6
[7] dir.expiry_1.12.0 DelayedMatrixStats_1.26.0
[9] png_0.1-8 vctrs_0.6.5
[11] pkgconfig_2.0.3 crayon_1.5.2
[13] fastmap_1.1.1 dbplyr_2.5.0
[15] magick_2.8.3 XVector_0.44.0
[17] utf8_1.2.4 rmarkdown_2.26
[19] graph_1.82.0 UCSC.utils_1.0.0
[21] purrr_1.0.2 bit_4.0.5
[23] xfun_0.43 bluster_1.14.0
[25] zlibbioc_1.50.0 cachem_1.0.8
[27] beachmat_2.20.0 jsonlite_1.8.8
[29] blob_1.2.4 highr_0.10
[31] rhdf5filters_1.16.0 DelayedArray_0.30.0
[33] Rhdf5lib_1.26.0 BiocParallel_1.38.0
[35] cluster_2.1.6 irlba_2.3.5.1
[37] parallel_4.4.0 R6_2.5.1
[39] bslib_0.7.0 jquerylib_0.1.4
[41] Rcpp_1.0.12 bookdown_0.39
[43] knitr_1.46 R.utils_2.12.3
[45] splines_4.4.0 igraph_2.0.3
[47] Matrix_1.7-0 tidyselect_1.2.1
[49] abind_1.4-5 yaml_2.3.8
[51] codetools_0.2-20 curl_5.2.1
[53] lattice_0.22-6 tibble_3.2.1
[55] withr_3.0.0 KEGGREST_1.44.0
[57] BumpyMatrix_1.12.0 evaluate_0.23
[59] BiocFileCache_2.12.0 ExperimentHub_2.12.0
[61] Biostrings_2.72.0 pillar_1.9.0
[63] BiocManager_1.30.22 filelock_1.0.3
[65] generics_0.1.3 BiocVersion_3.19.1
[67] sparseMatrixStats_1.16.0 glue_1.7.0
[69] metapod_1.12.0 tools_4.4.0
[71] AnnotationHub_3.12.0 BiocNeighbors_1.22.0
[73] ScaledMatrix_1.12.0 locfit_1.5-9.9
[75] XML_3.99-0.16.1 rhdf5_2.48.0
[77] grid_4.4.0 AnnotationDbi_1.66.0
[79] GenomeInfoDbData_1.2.12 HDF5Array_1.32.0
[81] BiocSingular_1.20.0 cli_3.6.2
[83] rsvd_1.0.5 rappdirs_0.3.3
[85] fansi_1.0.6 S4Arrays_1.4.0
[87] dplyr_1.1.4 R.methodsS3_1.8.2
[89] sass_0.4.9 digest_0.6.35
[91] dqrng_0.3.2 SparseArray_1.4.0
[93] rjson_0.2.21 R.oo_1.26.0
[95] memoise_2.0.1 htmltools_0.5.8.1
[97] lifecycle_1.0.4 httr_1.4.7
[99] statmod_1.5.0 mime_0.12
[101] bit64_4.0.5
```