---
title: "recount quick start guide"
author: "L Collado-Torres"
date: "`r doc_date()`"
package: "`r pkg_ver('recount')`"
output:
BiocStyle::html_document
vignette: >
%\VignetteIndexEntry{recount quick start guide}
%\VignetteEngine{knitr::rmarkdown}
%\VignetteEncoding{UTF-8}
---
# Basics
## Install `r Biocpkg('recount')`
`R` is an open-source statistical environment which can be easily modified to enhance its functionality via packages. `r Biocpkg('recount')` is a `R` package available via the [Bioconductor](http://bioconductor/packages/recount) repository for packages. `R` can be installed on any operating system from [CRAN](https://cran.r-project.org/) after which you can install `r Biocpkg('recount')` by using the following commands in your `R` session:
```{r 'installDer', eval = FALSE}
## try http:// if https:// URLs are not supported
source("https://bioconductor.org/biocLite.R")
biocLite("recount")
## Check that you have a valid Bioconductor installation
biocValid()
```
## Required knowledge
`r Biocpkg('recount')` is based on many other packages and in particular in those that have implemented the infrastructure needed for dealing with RNA-seq data. That is, packages like `r Biocpkg('GenomicFeatures')` and `r Biocpkg('rtracklayer')` that allow you to import the data. A `r Biocpkg('recount')` user is not expected to deal with those packages directly but will need to be familiar with `r Biocpkg('SummarizedExperiment')` to understand the results `r Biocpkg('recount')` generates. It might also prove to be highly beneficial to check the
* `r Biocpkg('DESeq2')` package for performing differential expression analysis with the _RangedSummarizedExperiment_ objects,
* `r Biocpkg('DEFormats')` package for switching the objects to those used by other differential expression packages such as `r Biocpkg('edgeR')`,
* `r Biocpkg('derfinder')` package for performing annotation-agnostic differential expression analysis.
If you are asking yourself the question "Where do I start using Bioconductor?" you might be interested in [this blog post](http://lcolladotor.github.io/2014/10/16/startBioC/#.VkOKbq6rRuU).
## Asking for help
As package developers, we try to explain clearly how to use our packages and in which order to use the functions. But `R` and `Bioconductor` have a steep learning curve so it is critical to learn where to ask for help. The blog post quoted above mentions some but we would like to highlight the [Bioconductor support site](https://support.bioconductor.org/) as the main resource for getting help: remember to use the `recount` tag and check [the older posts](https://support.bioconductor.org/t/recount/). Other alternatives are available such as creating GitHub issues and tweeting. However, please note that if you want to receive help you should adhere to the [posting guidelines](http://www.bioconductor.org/help/support/posting-guide/). It is particularly critical that you provide a small reproducible example and your session information so package developers can track down the source of the error.
## Citing `r Biocpkg('recount')`
We hope that `r Biocpkg('recount')` will be useful for your research. Please use the following information to cite the package and the overall approach. Thank you!
```{r 'citation'}
## Citation info
citation('recount')
```
# Quick start to using to `r Biocpkg('recount')`
__As of January 30, 2017 the annotation used for the exon and gene counts is Gencode v25.__
 ```{r vignetteSetup, echo=FALSE, message=FALSE, warning = FALSE}
## Track time spent on making the vignette
startTime <- Sys.time()
## Bib setup
library('knitcitations')
## Load knitcitations with a clean bibliography
cleanbib()
cite_options(hyperlink = 'to.doc', citation_format = 'text', style = 'html')
# Note links won't show for now due to the following issue
# https://github.com/cboettig/knitcitations/issues/63
## Write bibliography information
bibs <- c(
AnnotationDbi = citation('AnnotationDbi'),
BiocParallel = citation('BiocParallel'),
BiocStyle = citation('BiocStyle'),
derfinder = citation('derfinder')[1],
DESeq2 = citation('DESeq2'),
devtools = citation('devtools'),
downloader = citation('downloader'),
EnsDb.Hsapiens.v79 = citation('EnsDb.Hsapiens.v79'),
GEOquery = citation('GEOquery'),
GenomeInfoDb = citation('GenomeInfoDb'),
GenomicFeatures = citation('GenomicFeatures'),
GenomicRanges = citation('GenomicRanges'),
IRanges = citation('IRanges'),
knitcitations = citation('knitcitations'),
knitr = citation('knitr')[3],
org.Hs.eg.db = citation('org.Hs.eg.db'),
R = citation(),
RCurl = citation('RCurl'),
recount = citation('recount'),
regionReport = citation('regionReport'),
rentrez = citation('rentrez'),
rmarkdown = citation('rmarkdown'),
rtracklayer = citation('rtracklayer'),
S4Vectors = citation('S4Vectors'),
SummarizedExperiment = citation('SummarizedExperiment'),
testthat = citation('testthat')
)
write.bibtex(bibs,
file = 'quickstartRef.bib')
bib <- read.bibtex('quickstartRef.bib')
## Assign short names
names(bib) <- names(bibs)
```
Here is a very quick example of how to download a `RangedSummarizedExperiment` object with the gene counts for a 2 groups project (12 samples) with SRA study id [SRP009615](http://trace.ncbi.nlm.nih.gov/Traces/sra/?study=SRP009615) using the `r Biocpkg('recount')` package `r citep(bib[['recount']])`. The `RangedSummarizedExperiment` object is defined in the `r Biocpkg('SummarizedExperiment')` `r citep(bib[['SummarizedExperiment']])` package and can be used for differential expression analysis with different packages. Here we show how to use `r Biocpkg('DESeq2')` `r citep(bib[['DESeq2']])` to perform the differential expresion analysis.
This quick analysis is explained in more detail later on in this document. Further information about the recount project can be found in the publication [pre-print](http://biorxiv.org/content/early/2016/08/08/068478).
```{r 'ultraQuick', eval = FALSE}
## Load library
library('recount')
## Find a project of interest
project_info <- abstract_search('GSE32465')
## Download the gene-level RangedSummarizedExperiment data
download_study(project_info$project)
## Load the data
load(file.path(project_info$project, 'rse_gene.Rdata'))
## Browse the project at SRA
browse_study(project_info$project)
## View GEO ids
colData(rse_gene)$geo_accession
## Extract the sample characteristics
geochar <- lapply(split(colData(rse_gene), seq_len(nrow(colData(rse_gene)))), geo_characteristics)
## Note that the information for this study is a little inconsistent, so we
## have to fix it.
geochar <- do.call(rbind, lapply(geochar, function(x) {
if('cells' %in% colnames(x)) {
colnames(x)[colnames(x) == 'cells'] <- 'cell.line'
return(x)
} else {
return(x)
}
}))
## We can now define some sample information to use
sample_info <- data.frame(
run = colData(rse_gene)$run,
group = ifelse(grepl('uninduced', colData(rse_gene)$title), 'uninduced', 'induced'),
gene_target = sapply(colData(rse_gene)$title, function(x) { strsplit(strsplit(x,
'targeting ')[[1]][2], ' gene')[[1]][1] }),
cell.line = geochar$cell.line
)
## Scale counts by taking into account the total coverage per sample
rse <- scale_counts(rse_gene)
## Add sample information for DE analysis
colData(rse)$group <- sample_info$group
colData(rse)$gene_target <- sample_info$gene_target
## Perform differential expression analysis with DESeq2
library('DESeq2')
## Specify design and switch to DESeq2 format
dds <- DESeqDataSet(rse, ~ gene_target + group)
## Perform DE analysis
dds <- DESeq(dds, test = 'LRT', reduced = ~ gene_target, fitType = 'local')
res <- results(dds)
## Explore results
plotMA(res, main="DESeq2 results for SRP009615")
## Make a report with the results
library('regionReport')
DESeq2Report(dds, res = res, project = 'SRP009615',
intgroup = c('group', 'gene_target'), outdir = '.',
output = 'SRP009615-results')
```
The [recount project](https://jhubiostatistics.shinyapps.io/recount/) also hosts the necessary data to perform annotation-agnostic differential expression analyses with `r Biocpkg('derfinder')` `r citep(bib[['derfinder']])`. An example analysis would like this:
```{r 'er_analysis', eval = FALSE}
## Define expressed regions for study SRP009615, only for chromosome Y
regions <- expressed_regions('SRP009615', 'chrY', cutoff = 5L,
maxClusterGap = 3000L)
## Compute coverage matrix for study SRP009615, only for chromosome Y
system.time( rse_ER <- coverage_matrix('SRP009615', 'chrY', regions) )
## Round the coverage matrix to integers
covMat <- round(assays(rse_ER)$counts, 0)
## Get phenotype data for study SRP009615
pheno <- colData(rse_ER)
## Complete the phenotype table with the data we got from GEO
m <- match(pheno$run, sample_info$run)
pheno <- cbind(pheno, sample_info[m, 2:3])
## Build a DESeqDataSet
dds_ers <- DESeqDataSetFromMatrix(countData = covMat, colData = pheno,
design = ~ gene_target + group)
## Perform differential expression analysis with DESeq2 at the ER-level
dds_ers <- DESeq(dds_ers, test = 'LRT', reduced = ~ gene_target,
fitType = 'local')
res_ers <- results(dds_ers)
## Explore results
plotMA(res_ers, main="DESeq2 results for SRP009615 (ER-level, chrY)")
## Create a more extensive exploratory report
DESeq2Report(dds_ers, res = res_ers,
project = 'SRP009615 (ER-level, chrY)',
intgroup = c('group', 'gene_target'), outdir = '.',
output = 'SRP009615-results-ER-level-chrY')
```
# Introduction
`r Biocpkg('recount')` is an R package that provides an interface to the [recount project website](https://jhubiostatistics.shinyapps.io/recount/). This package allows you to download the files from the recount project and has helper functions for getting you started with differential expression analyses. This vignette will walk you through an example.
# Sample DE analysis
This is a brief overview of what you can do with `r Biocpkg('recount')`. In this particular example we will download data from the [SRP009615](http://trace.ncbi.nlm.nih.gov/Traces/sra/?study=SRP009615) study which sequenced 12 samples as described in the previous link.
If you don't have `r Biocpkg('recount')` installed, please do so with:
```{r 'install', eval = FALSE}
## try http:// if https:// URLs are not supported
source("https://bioconductor.org/biocLite.R")
biocLite("recount")
```
Next we load the required packages. Loading `r Biocpkg('recount')` will load the required dependencies.
```{r 'start', message=FALSE}
## Load recount R package
library('recount')
```
Lets say that we don't know the actual SRA accession number for this study but we do know a particular term which will help us identify it. If that's the case, we can use the `abstract_search()` function to identify the study of interest as shown below.
```{r 'search_abstract'}
## Find a project of interest
project_info <- abstract_search('GSE32465')
## Explore info
project_info
```
## Download data
Now that we have a study that we are interested in, we can download the _RangedSummarizedExperiment_ object (see `r Biocpkg('SummarizedExperiment')`) with the data summarized at the gene level. The function `download_study()` helps us do this. If you are interested on how the annotation was defined, check `reproduce_ranges()` described in the _Annotation_ section further down.
```{r 'download'}
## Download the gene-level RangedSummarizedExperiment data
download_study(project_info$project)
## Load the data
load(file.path(project_info$project, 'rse_gene.Rdata'))
```
We can explore a bit this _RangedSummarizedExperiment_ as shown below.
```{r 'explore_rse'}
rse_gene
## This is the sample phenotype data provided by the recount project
colData(rse_gene)
## At the gene level, the row data includes the gene Gencode ids, the gene
## symbols and the sum of the reduced exons widths, which can be used for
## taking into account the gene length.
rowData(rse_gene)
## At the exon level, you can get the gene Gencode ids from the names of:
# rowRanges(rse_exon)
```
## Finding phenotype information
Once we have identified the study of interest, we can use the `browse_study()` function to browse the study at the SRA website.
```{r 'browse'}
## Browse the project at SRA
browse_study(project_info$project)
```
The SRA website includes an _Experiments_ link which further describes each of the samples. From the information available for [SRP009615 at NCBI](http://www.ncbi.nlm.nih.gov/sra/?term=SRP009615) we have some further sample information that we can save for use in our differential expression analysis. We can get some of this information from [GEO](http://www.ncbi.nlm.nih.gov/geo/). The function `find_geo()` finds the [GEO](http://www.ncbi.nlm.nih.gov/geo/) accession id for a given SRA run accession id, which we can then use with `geo_info()` and `geo_characteristics()` to parse this information. The `rse_gene` object already has some of this information.
```{r 'sample_info', warning = FALSE}
## View GEO ids
colData(rse_gene)$geo_accession
## Extract the sample characteristics
geochar <- lapply(split(colData(rse_gene), seq_len(nrow(colData(rse_gene)))), geo_characteristics)
## Note that the information for this study is a little inconsistent, so we
## have to fix it.
geochar <- do.call(rbind, lapply(geochar, function(x) {
if('cells' %in% colnames(x)) {
colnames(x)[colnames(x) == 'cells'] <- 'cell.line'
return(x)
} else {
return(x)
}
}))
## We can now define some sample information to use
sample_info <- data.frame(
run = colData(rse_gene)$run,
group = ifelse(grepl('uninduced', colData(rse_gene)$title), 'uninduced', 'induced'),
gene_target = sapply(colData(rse_gene)$title, function(x) { strsplit(strsplit(x,
'targeting ')[[1]][2], ' gene')[[1]][1] }),
cell.line = geochar$cell.line
)
```
## DE analysis
The [recount project](https://jhubiostatistics.shinyapps.io/recount/) records the sum of the base level coverage for each gene (or exon). These raw counts have to be scaled and there are several ways in which you can choose to do so. The function `scale_counts()` helps you scale them in a way that is tailored to [Rail-RNA](http://rail.bio) output.
```{r 'scale_counts'}
## Scale counts by taking into account the total coverage per sample
rse <- scale_counts(rse_gene)
```
We are almost ready to perform our differential expression analysis. Lets just add the information we recovered [GEO](http://www.ncbi.nlm.nih.gov/geo/) about these samples.
```{r 'add_sample_info'}
## Add sample information for DE analysis
colData(rse)$group <- sample_info$group
colData(rse)$gene_target <- sample_info$gene_target
```
Now that the _RangedSummarizedExperiment_ is complete, we can use `r Biocpkg('DESeq2')` or another package to perform the differential expression test. Note that you can use `r Biocpkg('DEFormats')` for switching between formats if you want to use another package, like `r Biocpkg('edgeR')`.
In this particular analysis, we'll test whether there is a group difference adjusting for the gene target.
```{r 'de_analysis'}
## Perform differential expression analysis with DESeq2
library('DESeq2')
## Specify design and switch to DESeq2 format
dds <- DESeqDataSet(rse, ~ gene_target + group)
## Perform DE analysis
dds <- DESeq(dds, test = 'LRT', reduced = ~ gene_target, fitType = 'local')
res <- results(dds)
```
We can now use functions from `r Biocpkg('DESeq2')` to explore the results. For more details check the `r Biocpkg('DESeq2')` vignette. For example, we can make a MA plot as shown below.
```{r 'ma_plot'}
## Explore results
plotMA(res, main="DESeq2 results for SRP009615")
```
## Report results
We can also use the `r Biocpkg('regionReport')` package to generate interactive HTML reports exploring the `r Biocpkg('DESeq2')` results (or `r Biocpkg('edgeR')` results if you used that package).
```{r 'make_report', eval = FALSE}
## Make a report with the results
library('regionReport')
report <- DESeq2Report(dds, res = res, project = 'SRP009615',
intgroup = c('group', 'gene_target'), outdir = '.',
output = 'SRP009615-results', nBest = 10, nBestFeatures = 2)
```
```{r 'make_report_real', echo = FALSE, results = 'hide'}
library('regionReport')
## Make it so that the report will be available as a vignette
original <- readLines(system.file('DESeq2Exploration', 'DESeq2Exploration.Rmd',
package = 'regionReport'))
vignetteInfo <- c(
'vignette: >',
' %\\VignetteEngine{knitr::rmarkdown}',
' %\\VignetteIndexEntry{Basic DESeq2 results exploration}',
' %\\VignetteEncoding{UTF-8}'
)
new <- c(original[1:16], vignetteInfo, original[17:length(original)])
writeLines(new, 'SRP009615-results-template.Rmd')
## Now create the report
report <- DESeq2Report(dds, res = res, project = 'SRP009615',
intgroup = c('group', 'gene_target'), outdir = '.',
output = 'SRP009615-results', device = 'png', template = 'SRP009615-results-template.Rmd', nBest = 10, nBestFeatures = 2)
## Clean up
file.remove('SRP009615-results-template.Rmd')
```
You can view the final report [here](SRP009615-results.html).
## Finding gene names
The gene Gencode ids are included in several objects in `recount`. With these ids you can find the gene symbols, gene names and other information. The `r Biocpkg('org.Hs.eg.db')` is useful for finding this information and the following code shows how to get the gene names and symbols.
```{r 'geneSymbols'}
## Load required library
library('org.Hs.eg.db')
## Extract Gencode gene ids
gencode <- gsub('\\..*', '', names(recount_genes))
## Find the gene information we are interested in
gene_info <- select(org.Hs.eg.db, gencode, c('ENTREZID', 'GENENAME', 'SYMBOL',
'ENSEMBL'), 'ENSEMBL')
## Explore part of the results
dim(gene_info)
head(gene_info)
```
# Sample `r Biocpkg('derfinder')` analysis
The [recount project](https://jhubiostatistics.shinyapps.io/recount/) also hosts for each project sample coverage [bigWig files](https://genome.ucsc.edu/goldenpath/help/bigWig.html) created by [Rail-RNA](http://rail.bio) and a mean coverage bigWig file. For the mean coverage bigWig file, all samples were normalized to libraries of 40 million reads, each a 100 base-pairs long. `r Biocpkg('recount')` can be used along with `r Biocpkg('derfinder')` `r citep(bib[['derfinder']])` to identify expressed regions from the data. This type of analysis is annotation-agnostic which can be advantageous. The following subsections illustrate this type of analysis.
## Define expressed regions
For an annotation-agnostic differential expression analysis, we first need to define the regions of interest. With `r Biocpkg('recount')` we can do so using the `expressed_regions()` function as shown below for the same study we studied earlier.
```{r 'define_ers', eval = .Platform$OS.type != 'windows'}
## Define expressed regions for study SRP009615, only for chromosome Y
regions <- expressed_regions('SRP009615', 'chrY', cutoff = 5L,
maxClusterGap = 3000L)
## Briefly explore the resulting regions
regions
```
Once the regions have been defined, you can export them into a BED file using `r Biocpkg('rtracklayer')` or other file formats.
## Compute coverage matrix
Having defined the expressed regions, we can now compute a coverage matrix for these regions. We can do so using the function `coverage_matrix()` from `r Biocpkg('recount')` as shown below. It returns a _RangedSummarizedExperiment_ object.
```{r 'compute_covMat', eval = .Platform$OS.type != 'windows'}
## Compute coverage matrix for study SRP009615, only for chromosome Y
system.time( rse_ER <- coverage_matrix('SRP009615', 'chrY', regions) )
## Explore the RSE a bit
dim(rse_ER)
rse_ER
```
The resulting count matrix has one row per region and one column per sample. The counts correspond to the number (or fraction) of reads overlapping the regions. For some differential expression methods, you might have to round this matrix into integers. We'll use `r Biocpkg('DESeq2')` to identify which expressed regions are differentially expressed.
```{r 'to_integer', eval = .Platform$OS.type != 'windows'}
## Round the coverage matrix to integers
covMat <- round(assays(rse_ER)$counts, 0)
```
## Construct a `DESeqDataSet` object
We first need to get some phenotype information for these samples similar to the first analysis we did. We can get this data using `download_study()`.
```{r 'phenoData', eval = .Platform$OS.type != 'windows'}
## Get phenotype data for study SRP009615
pheno <- colData(rse_ER)
## Complete the phenotype table with the data we got from GEO
m <- match(pheno$run, sample_info$run)
pheno <- cbind(pheno, sample_info[m, 2:3])
## Explore the phenotype data a little bit
head(pheno)
```
Now that we have the necessary data, we can construct a `DESeqDataSet` object using the function `DESeqDataSetFromMatrix()` from `r Biocpkg('DESeq2')`.
```{r 'ers_dds', eval = .Platform$OS.type != 'windows'}
## Build a DESeqDataSet
dds_ers <- DESeqDataSetFromMatrix(countData = covMat, colData = pheno,
design = ~ gene_target + group)
```
## `r Biocpkg('DESeq2')` analysis
With the `DESeqDataSet` object in place, we can then use the function `DESeq()` from `r Biocpkg('DESeq2')` to perform the differential expression analysis (between groups) as shown below.
```{r 'de_analysis_ers', eval = .Platform$OS.type != 'windows'}
## Perform differential expression analysis with DESeq2 at the ER-level
dds_ers <- DESeq(dds_ers, test = 'LRT', reduced = ~ gene_target,
fitType = 'local')
res_ers <- results(dds_ers)
```
We can then visually explore the results, like we did before.
```{r 'ma_plot_ers', eval = .Platform$OS.type != 'windows'}
## Explore results
plotMA(res_ers, main="DESeq2 results for SRP009615 (ER-level, chrY)")
```
We can also use `r Biocpkg('regionReport')` to create a more extensive exploratory report.
```{r 'report2', eval = FALSE}
## Create the report
report2 <- DESeq2Report(dds_ers, res = res_ers,
project = 'SRP009615 (ER-level, chrY)',
intgroup = c('group', 'gene_target'), outdir = '.',
output = 'SRP009615-results-ER-level-chrY')
```
# Annotation used
This section describes the annotation used in `recount`.
We used the comprehensive gene annotation for (`CHR` regions) from [Gencode v25](https://www.gencodegenes.org/releases/25.html) (GRCh38.p7) to get the list of genes. Specifically [this GFF3](ftp://ftp.sanger.ac.uk/pub/gencode/Gencode_human/release_25/gencode.v25.annotation.gff3.gz) file. We then used the `r Biocpkg('org.Hs.eg.db')` package to get the gene symbols. For each gene, we also counted the total length of exonic base pairs for that given gene and stored this information in the `bp_length` column. You can see below this information for the `rse_gene` object included in `r Biocpkg('recount')`. The `rownames()` are the Gencode `gene_id`.
```{r, 'gene_annotation'}
## Gene annotation information
rowRanges(rse_gene_SRP009615)
## Also accessible via
rowData(rse_gene_SRP009615)
```
For an `rse_exon` object, the `rownames()` correspond to the Gencode `gene_id`. You can add a `gene_id` column if you are interested in subsetting the whole `rse_exon` object.
```{r, 'exon_annotation'}
## Get the rse_exon object for study SRP009615
download_study('SRP009615', type = 'rse-exon')
load(file.path('SRP009615', 'rse_exon.Rdata'))
## Annotation information
rowRanges(rse_exon)
## Add a gene_id column
rowRanges(rse_exon)$gene_id <- rownames(rse_exon)
## Example subset
rse_exon_subset <- subset(rse_exon, subset = gene_id == 'ENSG00000000003.14')
rowRanges(rse_exon_subset)
```
The exons in a `rse_exon` object are those that have a Gencode `gene_id` and that were reduced as described in the `r Biocpkg('GenomicRanges')` package:
```
?`inter-range-methods`
```
You can reproduce the gene and exon information using the function `reproduce_ranges()`.
## Using another/newer annotation
If you are interested in using another annotation based on hg38 coordinates you can do so using the function `coverage_matrix()` or by running [bwtool](https://github.com/CRG-Barcelona/bwtool/wiki) thanks to the BigWig files in `recount`, in which case [recount.bwtool](https://github.com/LieberInstitute/recount.bwtool) will be helpful.
If you are re-processing a small set of samples, it simply might be easier to use `coverage_matrix()` as shown below using the annotation information provided by the `r Biocpkg('EnsDb.Hsapiens.v79')` package.
```{r, eval = .Platform$OS.type != 'windows'}
## Get the reduced exons based on EnsDb.Hsapiens.v79 which matches hg38
exons <- reproduce_ranges('exon', db = 'EnsDb.Hsapiens.v79')
## Change the chromosome names to match those used in the BigWig files
library('GenomeInfoDb')
seqlevelsStyle(exons) <- 'UCSC'
## Get the count matrix at the exon level for chrY
exons_chrY <- keepSeqlevels(exons, 'chrY')
exonRSE <- coverage_matrix('SRP009615', 'chrY', unlist(exons_chrY),
chunksize = 3000)
exonMatrix <- assays(exonRSE)$counts
dim(exonMatrix)
head(exonMatrix)
## Summary the information at the gene level for chrY
exon_gene <- rep(names(exons_chrY), elementNROWS(exons_chrY))
geneMatrix <- do.call(rbind, lapply(split(as.data.frame(exonMatrix),
exon_gene), colSums))
dim(geneMatrix)
head(geneMatrix)
```
The above solution works well for a small set of samples. However, `bwtool` is faster for processing large sets. You can see how we used `bwtool` at [leekgroup/recount-website](https://github.com/leekgroup/recount-website). It's much faster than R and the small package [recount.bwtool](https://github.com/LieberInstitute/recount.bwtool) will be helpful for such scenarios. Note that you will need to run `scale_counts()` after using `coverage_matrix_bwtool()` and that will have to download the BigWig files first unless you are working at [JHPCE](https://jhpce.jhu.edu/) or [SciServer](http://www.sciserver.org/). `coverage_matrix_bwtool()` will download the files for you, but that till take time.
Finally, the BigWig files hosted by recount have the following chromosome names and sizes as specified in [hg38.sizes](https://github.com/nellore/runs/blob/master/gtex/hg38.sizes). You'll have to make sure that you match these names when using alternative annotations.
# Candidate gene fusions
If you are interested in finding possible gene fusion events in a particular study, you can download the _RangedSummarizedExperiment_ object at the exon-exon junctions level for that study. The objects we provide classify exon-exon junction events as shown below.
```{r 'gene_fusions'}
library('recount')
## Download and load RSE object for SRP009615 study
download_study('SRP009615', type = 'rse-jx')
load(file.path('SRP009615', 'rse_jx.Rdata'))
## Exon-exon junctions by class
table(rowRanges(rse_jx)$class)
## Potential gene fusions by chromosome
fusions <- rowRanges(rse_jx)[rowRanges(rse_jx)$class == 'fusion']
fusions_by_chr <- sort(table(seqnames(fusions)), decreasing = TRUE)
fusions_by_chr[fusions_by_chr > 0]
## Genes with the most fusions
head(sort(table(unlist(fusions$symbol_proposed)), decreasing = TRUE))
```
If you are interested in checking the frequency of a particular exon-exon junction then check the `snaptron_query()` function described in the following section.
# Snaptron
Our collaborators built _SnaptronUI_ to query the [Intropolis](https://github.com/nellore/intropolis) database of the exon-exon junctions found in SRA. _SnaptronUI_ allows you to do several types of queries manually and perform analyses. `r Biocpkg('recount')` has the function `snaptron_query()` which uses [Snaptron](https://github.com/ChristopherWilks/snaptron) to check if particular exon-exon junctions are present in _Intropolis_. For example, here we use `snaptron_query()` to get in `R`-friendly format the information for 3 exon-exon junctions using _Intropolis_ version 1 which uses hg19 coordinates. Version 2 uses hg38 coordinates and the exon-exon junctions were derived from a larger data set: about 50,000 samples vs 21,000 in version 1. `snaptron_query()` can also be used to access the 30 million and 36 million exon-exon junctions derived from the GTEx and TCGA consortiums respectively (about 10 and 11 thousand samples respectively).
```{r snaptron}
library('GenomicRanges')
junctions <- GRanges(seqnames = 'chr2', IRanges(
start = c(28971711, 29555082, 29754983),
end = c(29462418, 29923339, 29917715)))
snaptron_query(junctions)
```
If you use `snaptron_query()` please cite [snaptron.cs.jhu.edu/snaptron/docs/](http://snaptron.cs.jhu.edu/snaptron/docs/). _Snaptron_ is separate from `r Biocpkg('recount')`. Thank you!
# Download all the data
If you are interested in downloading all the data you can do so using the `recount_url` data.frame included in the package. That is:
```{r 'downloadAll'}
## Get data.frame with all the URLs
library('recount')
dim(recount_url)
## Explore URLs
head(recount_url$url)
```
You can then download all the data using the `download_study()` function or however you prefer to do so with `recount_url`.
```{r 'actuallyDownload', eval = FALSE}
## Download all files (will take a while! It's over 8 TB of data)
sapply(unique(recount_url$project), download_study, type = 'all')
```
You can find the code for the website at [leekgroup/recount-website](https://github.com/leekgroup/recount-website/tree/master/website) if you want to deploy your own local mirror. Please let us know if you choose to do deploy a mirror.
# Accessing `r Biocpkg('recount')` via _SciServer_
Not everyone has over 8 terabytes of disk space available to download all the data from the `recount` project. However, thanks to [SciServer](http://www.sciserver.org/) you can access it locally via a Jupyter Notebook. If you do so and want to share your work, please let the [SciServer](http://www.sciserver.org/) maintainers know via Twitter at [IDIESJHU](https://twitter.com/IDIESJHU).
## Step by step
To use _SciServer_, you first have to go to http://www.sciserver.org/ then click on "Login to SciServer". If you are a first time user, click on "Register New Account" and enter the information they request as shown on the next image.
```{r vignetteSetup, echo=FALSE, message=FALSE, warning = FALSE}
## Track time spent on making the vignette
startTime <- Sys.time()
## Bib setup
library('knitcitations')
## Load knitcitations with a clean bibliography
cleanbib()
cite_options(hyperlink = 'to.doc', citation_format = 'text', style = 'html')
# Note links won't show for now due to the following issue
# https://github.com/cboettig/knitcitations/issues/63
## Write bibliography information
bibs <- c(
AnnotationDbi = citation('AnnotationDbi'),
BiocParallel = citation('BiocParallel'),
BiocStyle = citation('BiocStyle'),
derfinder = citation('derfinder')[1],
DESeq2 = citation('DESeq2'),
devtools = citation('devtools'),
downloader = citation('downloader'),
EnsDb.Hsapiens.v79 = citation('EnsDb.Hsapiens.v79'),
GEOquery = citation('GEOquery'),
GenomeInfoDb = citation('GenomeInfoDb'),
GenomicFeatures = citation('GenomicFeatures'),
GenomicRanges = citation('GenomicRanges'),
IRanges = citation('IRanges'),
knitcitations = citation('knitcitations'),
knitr = citation('knitr')[3],
org.Hs.eg.db = citation('org.Hs.eg.db'),
R = citation(),
RCurl = citation('RCurl'),
recount = citation('recount'),
regionReport = citation('regionReport'),
rentrez = citation('rentrez'),
rmarkdown = citation('rmarkdown'),
rtracklayer = citation('rtracklayer'),
S4Vectors = citation('S4Vectors'),
SummarizedExperiment = citation('SummarizedExperiment'),
testthat = citation('testthat')
)
write.bibtex(bibs,
file = 'quickstartRef.bib')
bib <- read.bibtex('quickstartRef.bib')
## Assign short names
names(bib) <- names(bibs)
```
Here is a very quick example of how to download a `RangedSummarizedExperiment` object with the gene counts for a 2 groups project (12 samples) with SRA study id [SRP009615](http://trace.ncbi.nlm.nih.gov/Traces/sra/?study=SRP009615) using the `r Biocpkg('recount')` package `r citep(bib[['recount']])`. The `RangedSummarizedExperiment` object is defined in the `r Biocpkg('SummarizedExperiment')` `r citep(bib[['SummarizedExperiment']])` package and can be used for differential expression analysis with different packages. Here we show how to use `r Biocpkg('DESeq2')` `r citep(bib[['DESeq2']])` to perform the differential expresion analysis.
This quick analysis is explained in more detail later on in this document. Further information about the recount project can be found in the publication [pre-print](http://biorxiv.org/content/early/2016/08/08/068478).
```{r 'ultraQuick', eval = FALSE}
## Load library
library('recount')
## Find a project of interest
project_info <- abstract_search('GSE32465')
## Download the gene-level RangedSummarizedExperiment data
download_study(project_info$project)
## Load the data
load(file.path(project_info$project, 'rse_gene.Rdata'))
## Browse the project at SRA
browse_study(project_info$project)
## View GEO ids
colData(rse_gene)$geo_accession
## Extract the sample characteristics
geochar <- lapply(split(colData(rse_gene), seq_len(nrow(colData(rse_gene)))), geo_characteristics)
## Note that the information for this study is a little inconsistent, so we
## have to fix it.
geochar <- do.call(rbind, lapply(geochar, function(x) {
if('cells' %in% colnames(x)) {
colnames(x)[colnames(x) == 'cells'] <- 'cell.line'
return(x)
} else {
return(x)
}
}))
## We can now define some sample information to use
sample_info <- data.frame(
run = colData(rse_gene)$run,
group = ifelse(grepl('uninduced', colData(rse_gene)$title), 'uninduced', 'induced'),
gene_target = sapply(colData(rse_gene)$title, function(x) { strsplit(strsplit(x,
'targeting ')[[1]][2], ' gene')[[1]][1] }),
cell.line = geochar$cell.line
)
## Scale counts by taking into account the total coverage per sample
rse <- scale_counts(rse_gene)
## Add sample information for DE analysis
colData(rse)$group <- sample_info$group
colData(rse)$gene_target <- sample_info$gene_target
## Perform differential expression analysis with DESeq2
library('DESeq2')
## Specify design and switch to DESeq2 format
dds <- DESeqDataSet(rse, ~ gene_target + group)
## Perform DE analysis
dds <- DESeq(dds, test = 'LRT', reduced = ~ gene_target, fitType = 'local')
res <- results(dds)
## Explore results
plotMA(res, main="DESeq2 results for SRP009615")
## Make a report with the results
library('regionReport')
DESeq2Report(dds, res = res, project = 'SRP009615',
intgroup = c('group', 'gene_target'), outdir = '.',
output = 'SRP009615-results')
```
The [recount project](https://jhubiostatistics.shinyapps.io/recount/) also hosts the necessary data to perform annotation-agnostic differential expression analyses with `r Biocpkg('derfinder')` `r citep(bib[['derfinder']])`. An example analysis would like this:
```{r 'er_analysis', eval = FALSE}
## Define expressed regions for study SRP009615, only for chromosome Y
regions <- expressed_regions('SRP009615', 'chrY', cutoff = 5L,
maxClusterGap = 3000L)
## Compute coverage matrix for study SRP009615, only for chromosome Y
system.time( rse_ER <- coverage_matrix('SRP009615', 'chrY', regions) )
## Round the coverage matrix to integers
covMat <- round(assays(rse_ER)$counts, 0)
## Get phenotype data for study SRP009615
pheno <- colData(rse_ER)
## Complete the phenotype table with the data we got from GEO
m <- match(pheno$run, sample_info$run)
pheno <- cbind(pheno, sample_info[m, 2:3])
## Build a DESeqDataSet
dds_ers <- DESeqDataSetFromMatrix(countData = covMat, colData = pheno,
design = ~ gene_target + group)
## Perform differential expression analysis with DESeq2 at the ER-level
dds_ers <- DESeq(dds_ers, test = 'LRT', reduced = ~ gene_target,
fitType = 'local')
res_ers <- results(dds_ers)
## Explore results
plotMA(res_ers, main="DESeq2 results for SRP009615 (ER-level, chrY)")
## Create a more extensive exploratory report
DESeq2Report(dds_ers, res = res_ers,
project = 'SRP009615 (ER-level, chrY)',
intgroup = c('group', 'gene_target'), outdir = '.',
output = 'SRP009615-results-ER-level-chrY')
```
# Introduction
`r Biocpkg('recount')` is an R package that provides an interface to the [recount project website](https://jhubiostatistics.shinyapps.io/recount/). This package allows you to download the files from the recount project and has helper functions for getting you started with differential expression analyses. This vignette will walk you through an example.
# Sample DE analysis
This is a brief overview of what you can do with `r Biocpkg('recount')`. In this particular example we will download data from the [SRP009615](http://trace.ncbi.nlm.nih.gov/Traces/sra/?study=SRP009615) study which sequenced 12 samples as described in the previous link.
If you don't have `r Biocpkg('recount')` installed, please do so with:
```{r 'install', eval = FALSE}
## try http:// if https:// URLs are not supported
source("https://bioconductor.org/biocLite.R")
biocLite("recount")
```
Next we load the required packages. Loading `r Biocpkg('recount')` will load the required dependencies.
```{r 'start', message=FALSE}
## Load recount R package
library('recount')
```
Lets say that we don't know the actual SRA accession number for this study but we do know a particular term which will help us identify it. If that's the case, we can use the `abstract_search()` function to identify the study of interest as shown below.
```{r 'search_abstract'}
## Find a project of interest
project_info <- abstract_search('GSE32465')
## Explore info
project_info
```
## Download data
Now that we have a study that we are interested in, we can download the _RangedSummarizedExperiment_ object (see `r Biocpkg('SummarizedExperiment')`) with the data summarized at the gene level. The function `download_study()` helps us do this. If you are interested on how the annotation was defined, check `reproduce_ranges()` described in the _Annotation_ section further down.
```{r 'download'}
## Download the gene-level RangedSummarizedExperiment data
download_study(project_info$project)
## Load the data
load(file.path(project_info$project, 'rse_gene.Rdata'))
```
We can explore a bit this _RangedSummarizedExperiment_ as shown below.
```{r 'explore_rse'}
rse_gene
## This is the sample phenotype data provided by the recount project
colData(rse_gene)
## At the gene level, the row data includes the gene Gencode ids, the gene
## symbols and the sum of the reduced exons widths, which can be used for
## taking into account the gene length.
rowData(rse_gene)
## At the exon level, you can get the gene Gencode ids from the names of:
# rowRanges(rse_exon)
```
## Finding phenotype information
Once we have identified the study of interest, we can use the `browse_study()` function to browse the study at the SRA website.
```{r 'browse'}
## Browse the project at SRA
browse_study(project_info$project)
```
The SRA website includes an _Experiments_ link which further describes each of the samples. From the information available for [SRP009615 at NCBI](http://www.ncbi.nlm.nih.gov/sra/?term=SRP009615) we have some further sample information that we can save for use in our differential expression analysis. We can get some of this information from [GEO](http://www.ncbi.nlm.nih.gov/geo/). The function `find_geo()` finds the [GEO](http://www.ncbi.nlm.nih.gov/geo/) accession id for a given SRA run accession id, which we can then use with `geo_info()` and `geo_characteristics()` to parse this information. The `rse_gene` object already has some of this information.
```{r 'sample_info', warning = FALSE}
## View GEO ids
colData(rse_gene)$geo_accession
## Extract the sample characteristics
geochar <- lapply(split(colData(rse_gene), seq_len(nrow(colData(rse_gene)))), geo_characteristics)
## Note that the information for this study is a little inconsistent, so we
## have to fix it.
geochar <- do.call(rbind, lapply(geochar, function(x) {
if('cells' %in% colnames(x)) {
colnames(x)[colnames(x) == 'cells'] <- 'cell.line'
return(x)
} else {
return(x)
}
}))
## We can now define some sample information to use
sample_info <- data.frame(
run = colData(rse_gene)$run,
group = ifelse(grepl('uninduced', colData(rse_gene)$title), 'uninduced', 'induced'),
gene_target = sapply(colData(rse_gene)$title, function(x) { strsplit(strsplit(x,
'targeting ')[[1]][2], ' gene')[[1]][1] }),
cell.line = geochar$cell.line
)
```
## DE analysis
The [recount project](https://jhubiostatistics.shinyapps.io/recount/) records the sum of the base level coverage for each gene (or exon). These raw counts have to be scaled and there are several ways in which you can choose to do so. The function `scale_counts()` helps you scale them in a way that is tailored to [Rail-RNA](http://rail.bio) output.
```{r 'scale_counts'}
## Scale counts by taking into account the total coverage per sample
rse <- scale_counts(rse_gene)
```
We are almost ready to perform our differential expression analysis. Lets just add the information we recovered [GEO](http://www.ncbi.nlm.nih.gov/geo/) about these samples.
```{r 'add_sample_info'}
## Add sample information for DE analysis
colData(rse)$group <- sample_info$group
colData(rse)$gene_target <- sample_info$gene_target
```
Now that the _RangedSummarizedExperiment_ is complete, we can use `r Biocpkg('DESeq2')` or another package to perform the differential expression test. Note that you can use `r Biocpkg('DEFormats')` for switching between formats if you want to use another package, like `r Biocpkg('edgeR')`.
In this particular analysis, we'll test whether there is a group difference adjusting for the gene target.
```{r 'de_analysis'}
## Perform differential expression analysis with DESeq2
library('DESeq2')
## Specify design and switch to DESeq2 format
dds <- DESeqDataSet(rse, ~ gene_target + group)
## Perform DE analysis
dds <- DESeq(dds, test = 'LRT', reduced = ~ gene_target, fitType = 'local')
res <- results(dds)
```
We can now use functions from `r Biocpkg('DESeq2')` to explore the results. For more details check the `r Biocpkg('DESeq2')` vignette. For example, we can make a MA plot as shown below.
```{r 'ma_plot'}
## Explore results
plotMA(res, main="DESeq2 results for SRP009615")
```
## Report results
We can also use the `r Biocpkg('regionReport')` package to generate interactive HTML reports exploring the `r Biocpkg('DESeq2')` results (or `r Biocpkg('edgeR')` results if you used that package).
```{r 'make_report', eval = FALSE}
## Make a report with the results
library('regionReport')
report <- DESeq2Report(dds, res = res, project = 'SRP009615',
intgroup = c('group', 'gene_target'), outdir = '.',
output = 'SRP009615-results', nBest = 10, nBestFeatures = 2)
```
```{r 'make_report_real', echo = FALSE, results = 'hide'}
library('regionReport')
## Make it so that the report will be available as a vignette
original <- readLines(system.file('DESeq2Exploration', 'DESeq2Exploration.Rmd',
package = 'regionReport'))
vignetteInfo <- c(
'vignette: >',
' %\\VignetteEngine{knitr::rmarkdown}',
' %\\VignetteIndexEntry{Basic DESeq2 results exploration}',
' %\\VignetteEncoding{UTF-8}'
)
new <- c(original[1:16], vignetteInfo, original[17:length(original)])
writeLines(new, 'SRP009615-results-template.Rmd')
## Now create the report
report <- DESeq2Report(dds, res = res, project = 'SRP009615',
intgroup = c('group', 'gene_target'), outdir = '.',
output = 'SRP009615-results', device = 'png', template = 'SRP009615-results-template.Rmd', nBest = 10, nBestFeatures = 2)
## Clean up
file.remove('SRP009615-results-template.Rmd')
```
You can view the final report [here](SRP009615-results.html).
## Finding gene names
The gene Gencode ids are included in several objects in `recount`. With these ids you can find the gene symbols, gene names and other information. The `r Biocpkg('org.Hs.eg.db')` is useful for finding this information and the following code shows how to get the gene names and symbols.
```{r 'geneSymbols'}
## Load required library
library('org.Hs.eg.db')
## Extract Gencode gene ids
gencode <- gsub('\\..*', '', names(recount_genes))
## Find the gene information we are interested in
gene_info <- select(org.Hs.eg.db, gencode, c('ENTREZID', 'GENENAME', 'SYMBOL',
'ENSEMBL'), 'ENSEMBL')
## Explore part of the results
dim(gene_info)
head(gene_info)
```
# Sample `r Biocpkg('derfinder')` analysis
The [recount project](https://jhubiostatistics.shinyapps.io/recount/) also hosts for each project sample coverage [bigWig files](https://genome.ucsc.edu/goldenpath/help/bigWig.html) created by [Rail-RNA](http://rail.bio) and a mean coverage bigWig file. For the mean coverage bigWig file, all samples were normalized to libraries of 40 million reads, each a 100 base-pairs long. `r Biocpkg('recount')` can be used along with `r Biocpkg('derfinder')` `r citep(bib[['derfinder']])` to identify expressed regions from the data. This type of analysis is annotation-agnostic which can be advantageous. The following subsections illustrate this type of analysis.
## Define expressed regions
For an annotation-agnostic differential expression analysis, we first need to define the regions of interest. With `r Biocpkg('recount')` we can do so using the `expressed_regions()` function as shown below for the same study we studied earlier.
```{r 'define_ers', eval = .Platform$OS.type != 'windows'}
## Define expressed regions for study SRP009615, only for chromosome Y
regions <- expressed_regions('SRP009615', 'chrY', cutoff = 5L,
maxClusterGap = 3000L)
## Briefly explore the resulting regions
regions
```
Once the regions have been defined, you can export them into a BED file using `r Biocpkg('rtracklayer')` or other file formats.
## Compute coverage matrix
Having defined the expressed regions, we can now compute a coverage matrix for these regions. We can do so using the function `coverage_matrix()` from `r Biocpkg('recount')` as shown below. It returns a _RangedSummarizedExperiment_ object.
```{r 'compute_covMat', eval = .Platform$OS.type != 'windows'}
## Compute coverage matrix for study SRP009615, only for chromosome Y
system.time( rse_ER <- coverage_matrix('SRP009615', 'chrY', regions) )
## Explore the RSE a bit
dim(rse_ER)
rse_ER
```
The resulting count matrix has one row per region and one column per sample. The counts correspond to the number (or fraction) of reads overlapping the regions. For some differential expression methods, you might have to round this matrix into integers. We'll use `r Biocpkg('DESeq2')` to identify which expressed regions are differentially expressed.
```{r 'to_integer', eval = .Platform$OS.type != 'windows'}
## Round the coverage matrix to integers
covMat <- round(assays(rse_ER)$counts, 0)
```
## Construct a `DESeqDataSet` object
We first need to get some phenotype information for these samples similar to the first analysis we did. We can get this data using `download_study()`.
```{r 'phenoData', eval = .Platform$OS.type != 'windows'}
## Get phenotype data for study SRP009615
pheno <- colData(rse_ER)
## Complete the phenotype table with the data we got from GEO
m <- match(pheno$run, sample_info$run)
pheno <- cbind(pheno, sample_info[m, 2:3])
## Explore the phenotype data a little bit
head(pheno)
```
Now that we have the necessary data, we can construct a `DESeqDataSet` object using the function `DESeqDataSetFromMatrix()` from `r Biocpkg('DESeq2')`.
```{r 'ers_dds', eval = .Platform$OS.type != 'windows'}
## Build a DESeqDataSet
dds_ers <- DESeqDataSetFromMatrix(countData = covMat, colData = pheno,
design = ~ gene_target + group)
```
## `r Biocpkg('DESeq2')` analysis
With the `DESeqDataSet` object in place, we can then use the function `DESeq()` from `r Biocpkg('DESeq2')` to perform the differential expression analysis (between groups) as shown below.
```{r 'de_analysis_ers', eval = .Platform$OS.type != 'windows'}
## Perform differential expression analysis with DESeq2 at the ER-level
dds_ers <- DESeq(dds_ers, test = 'LRT', reduced = ~ gene_target,
fitType = 'local')
res_ers <- results(dds_ers)
```
We can then visually explore the results, like we did before.
```{r 'ma_plot_ers', eval = .Platform$OS.type != 'windows'}
## Explore results
plotMA(res_ers, main="DESeq2 results for SRP009615 (ER-level, chrY)")
```
We can also use `r Biocpkg('regionReport')` to create a more extensive exploratory report.
```{r 'report2', eval = FALSE}
## Create the report
report2 <- DESeq2Report(dds_ers, res = res_ers,
project = 'SRP009615 (ER-level, chrY)',
intgroup = c('group', 'gene_target'), outdir = '.',
output = 'SRP009615-results-ER-level-chrY')
```
# Annotation used
This section describes the annotation used in `recount`.
We used the comprehensive gene annotation for (`CHR` regions) from [Gencode v25](https://www.gencodegenes.org/releases/25.html) (GRCh38.p7) to get the list of genes. Specifically [this GFF3](ftp://ftp.sanger.ac.uk/pub/gencode/Gencode_human/release_25/gencode.v25.annotation.gff3.gz) file. We then used the `r Biocpkg('org.Hs.eg.db')` package to get the gene symbols. For each gene, we also counted the total length of exonic base pairs for that given gene and stored this information in the `bp_length` column. You can see below this information for the `rse_gene` object included in `r Biocpkg('recount')`. The `rownames()` are the Gencode `gene_id`.
```{r, 'gene_annotation'}
## Gene annotation information
rowRanges(rse_gene_SRP009615)
## Also accessible via
rowData(rse_gene_SRP009615)
```
For an `rse_exon` object, the `rownames()` correspond to the Gencode `gene_id`. You can add a `gene_id` column if you are interested in subsetting the whole `rse_exon` object.
```{r, 'exon_annotation'}
## Get the rse_exon object for study SRP009615
download_study('SRP009615', type = 'rse-exon')
load(file.path('SRP009615', 'rse_exon.Rdata'))
## Annotation information
rowRanges(rse_exon)
## Add a gene_id column
rowRanges(rse_exon)$gene_id <- rownames(rse_exon)
## Example subset
rse_exon_subset <- subset(rse_exon, subset = gene_id == 'ENSG00000000003.14')
rowRanges(rse_exon_subset)
```
The exons in a `rse_exon` object are those that have a Gencode `gene_id` and that were reduced as described in the `r Biocpkg('GenomicRanges')` package:
```
?`inter-range-methods`
```
You can reproduce the gene and exon information using the function `reproduce_ranges()`.
## Using another/newer annotation
If you are interested in using another annotation based on hg38 coordinates you can do so using the function `coverage_matrix()` or by running [bwtool](https://github.com/CRG-Barcelona/bwtool/wiki) thanks to the BigWig files in `recount`, in which case [recount.bwtool](https://github.com/LieberInstitute/recount.bwtool) will be helpful.
If you are re-processing a small set of samples, it simply might be easier to use `coverage_matrix()` as shown below using the annotation information provided by the `r Biocpkg('EnsDb.Hsapiens.v79')` package.
```{r, eval = .Platform$OS.type != 'windows'}
## Get the reduced exons based on EnsDb.Hsapiens.v79 which matches hg38
exons <- reproduce_ranges('exon', db = 'EnsDb.Hsapiens.v79')
## Change the chromosome names to match those used in the BigWig files
library('GenomeInfoDb')
seqlevelsStyle(exons) <- 'UCSC'
## Get the count matrix at the exon level for chrY
exons_chrY <- keepSeqlevels(exons, 'chrY')
exonRSE <- coverage_matrix('SRP009615', 'chrY', unlist(exons_chrY),
chunksize = 3000)
exonMatrix <- assays(exonRSE)$counts
dim(exonMatrix)
head(exonMatrix)
## Summary the information at the gene level for chrY
exon_gene <- rep(names(exons_chrY), elementNROWS(exons_chrY))
geneMatrix <- do.call(rbind, lapply(split(as.data.frame(exonMatrix),
exon_gene), colSums))
dim(geneMatrix)
head(geneMatrix)
```
The above solution works well for a small set of samples. However, `bwtool` is faster for processing large sets. You can see how we used `bwtool` at [leekgroup/recount-website](https://github.com/leekgroup/recount-website). It's much faster than R and the small package [recount.bwtool](https://github.com/LieberInstitute/recount.bwtool) will be helpful for such scenarios. Note that you will need to run `scale_counts()` after using `coverage_matrix_bwtool()` and that will have to download the BigWig files first unless you are working at [JHPCE](https://jhpce.jhu.edu/) or [SciServer](http://www.sciserver.org/). `coverage_matrix_bwtool()` will download the files for you, but that till take time.
Finally, the BigWig files hosted by recount have the following chromosome names and sizes as specified in [hg38.sizes](https://github.com/nellore/runs/blob/master/gtex/hg38.sizes). You'll have to make sure that you match these names when using alternative annotations.
# Candidate gene fusions
If you are interested in finding possible gene fusion events in a particular study, you can download the _RangedSummarizedExperiment_ object at the exon-exon junctions level for that study. The objects we provide classify exon-exon junction events as shown below.
```{r 'gene_fusions'}
library('recount')
## Download and load RSE object for SRP009615 study
download_study('SRP009615', type = 'rse-jx')
load(file.path('SRP009615', 'rse_jx.Rdata'))
## Exon-exon junctions by class
table(rowRanges(rse_jx)$class)
## Potential gene fusions by chromosome
fusions <- rowRanges(rse_jx)[rowRanges(rse_jx)$class == 'fusion']
fusions_by_chr <- sort(table(seqnames(fusions)), decreasing = TRUE)
fusions_by_chr[fusions_by_chr > 0]
## Genes with the most fusions
head(sort(table(unlist(fusions$symbol_proposed)), decreasing = TRUE))
```
If you are interested in checking the frequency of a particular exon-exon junction then check the `snaptron_query()` function described in the following section.
# Snaptron
Our collaborators built _SnaptronUI_ to query the [Intropolis](https://github.com/nellore/intropolis) database of the exon-exon junctions found in SRA. _SnaptronUI_ allows you to do several types of queries manually and perform analyses. `r Biocpkg('recount')` has the function `snaptron_query()` which uses [Snaptron](https://github.com/ChristopherWilks/snaptron) to check if particular exon-exon junctions are present in _Intropolis_. For example, here we use `snaptron_query()` to get in `R`-friendly format the information for 3 exon-exon junctions using _Intropolis_ version 1 which uses hg19 coordinates. Version 2 uses hg38 coordinates and the exon-exon junctions were derived from a larger data set: about 50,000 samples vs 21,000 in version 1. `snaptron_query()` can also be used to access the 30 million and 36 million exon-exon junctions derived from the GTEx and TCGA consortiums respectively (about 10 and 11 thousand samples respectively).
```{r snaptron}
library('GenomicRanges')
junctions <- GRanges(seqnames = 'chr2', IRanges(
start = c(28971711, 29555082, 29754983),
end = c(29462418, 29923339, 29917715)))
snaptron_query(junctions)
```
If you use `snaptron_query()` please cite [snaptron.cs.jhu.edu/snaptron/docs/](http://snaptron.cs.jhu.edu/snaptron/docs/). _Snaptron_ is separate from `r Biocpkg('recount')`. Thank you!
# Download all the data
If you are interested in downloading all the data you can do so using the `recount_url` data.frame included in the package. That is:
```{r 'downloadAll'}
## Get data.frame with all the URLs
library('recount')
dim(recount_url)
## Explore URLs
head(recount_url$url)
```
You can then download all the data using the `download_study()` function or however you prefer to do so with `recount_url`.
```{r 'actuallyDownload', eval = FALSE}
## Download all files (will take a while! It's over 8 TB of data)
sapply(unique(recount_url$project), download_study, type = 'all')
```
You can find the code for the website at [leekgroup/recount-website](https://github.com/leekgroup/recount-website/tree/master/website) if you want to deploy your own local mirror. Please let us know if you choose to do deploy a mirror.
# Accessing `r Biocpkg('recount')` via _SciServer_
Not everyone has over 8 terabytes of disk space available to download all the data from the `recount` project. However, thanks to [SciServer](http://www.sciserver.org/) you can access it locally via a Jupyter Notebook. If you do so and want to share your work, please let the [SciServer](http://www.sciserver.org/) maintainers know via Twitter at [IDIESJHU](https://twitter.com/IDIESJHU).
## Step by step
To use _SciServer_, you first have to go to http://www.sciserver.org/ then click on "Login to SciServer". If you are a first time user, click on "Register New Account" and enter the information they request as shown on the next image.
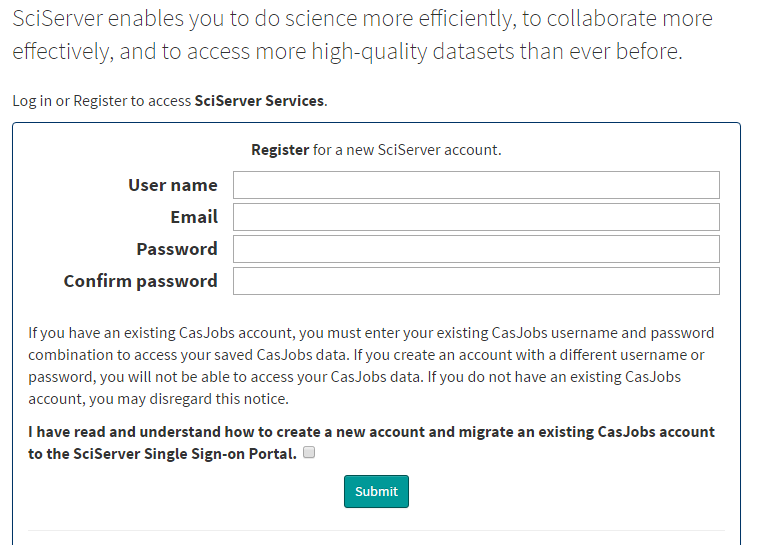 Once you've registered, open the [SciServer compute tool](http://compute.sciserver.org/dashboard/).
Once you've registered, open the [SciServer compute tool](http://compute.sciserver.org/dashboard/).
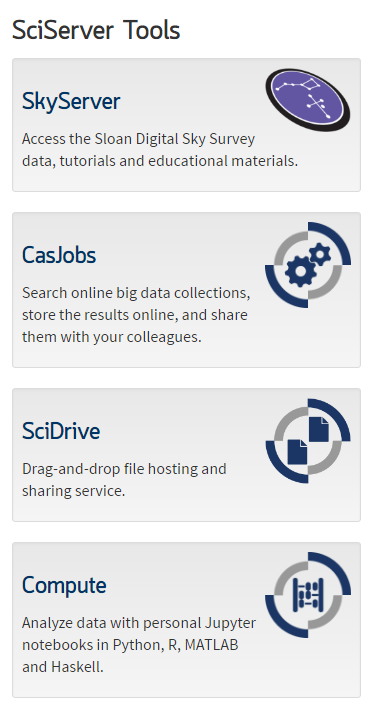 Then click on "Create container", choose a name of your preference and make sure that you choose to load R 3.3.x and the `recount` public volume. This will enable you to access all of the `recount` data as if it was on your computer. Click "Create" once you are ready.
Then click on "Create container", choose a name of your preference and make sure that you choose to load R 3.3.x and the `recount` public volume. This will enable you to access all of the `recount` data as if it was on your computer. Click "Create" once you are ready.
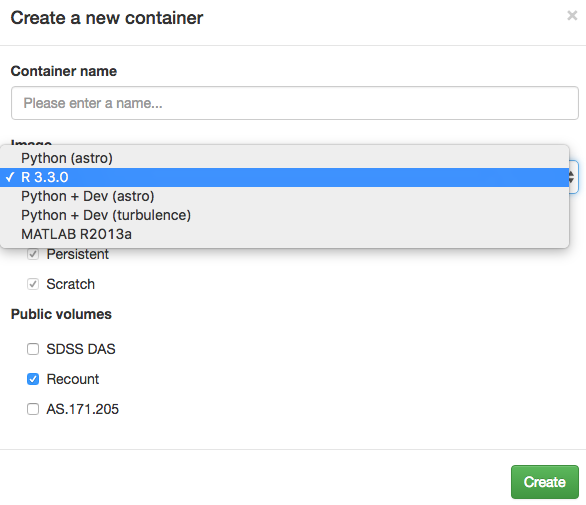 Once you have a container, create a new R _Notebook_ as shown in the image below.
Once you have a container, create a new R _Notebook_ as shown in the image below.
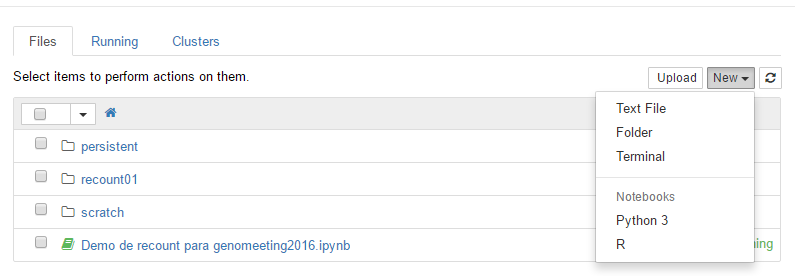 In this R _Notebook_ you can insert new code _cells_ where you can type R code. For example, you can install `r Biocpkg('recount')` and `r Biocpkg('DESeq2')` to run the code example from this vignette. You first have to install the packages as shown below.
In this R _Notebook_ you can insert new code _cells_ where you can type R code. For example, you can install `r Biocpkg('recount')` and `r Biocpkg('DESeq2')` to run the code example from this vignette. You first have to install the packages as shown below.
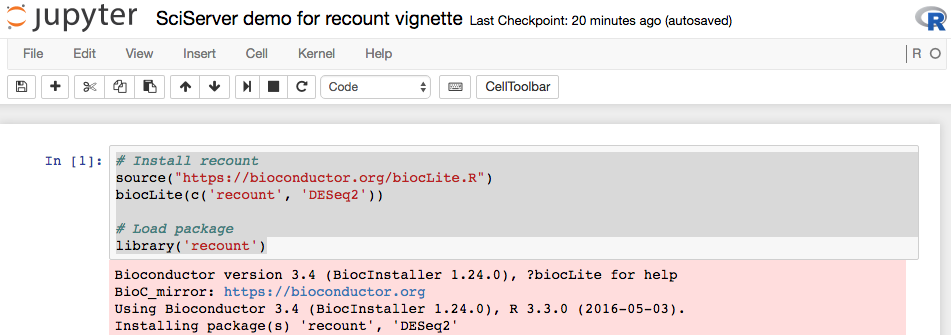 Then, the main part when using _SciServer_ is to use to remember that all the `recount` data is available locally. So instead of using `download_study()`, you can simply use `load()` with the correct path as shown below.
Then, the main part when using _SciServer_ is to use to remember that all the `recount` data is available locally. So instead of using `download_study()`, you can simply use `load()` with the correct path as shown below.
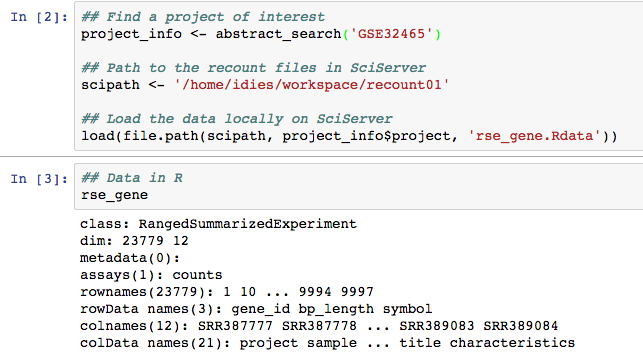 Several functions in the `r Biocpkg('recount')` package have the `outdir` argument, which you can specify to use the data locally. Try using `expressed_regions()` in _SciServer_ to get started with the following code:
```{r 'ER-sciserver', eval = FALSE}
## Expressed-regions SciServer example
regions <- expressed_regions('SRP009615', 'chrY', cutoff = 5L,
maxClusterGap = 3000L, outdir = file.path(scipath, 'SRP009615'))
regions
```
You can find the R code for a full _SciServer_ demo [here](https://github.com/leekgroup/recount-website/blob/master/sciserver_demo/sciserver_demo.R). You can copy and paste it into a cell and run it all to see how _SciServer compute_ works.
## SciServer help
If you encounter problems when using SciServer please check their [support page](http://www.sciserver.org/support/help/) and the [SciServer compute help section](http://compute.sciserver.org/dashboard/Home/Help).
## Citing _SciServer_
If you use [SciServer](http://www.sciserver.org/) for your published work, please cite it accordingly. _SciServer_ is administered by the [Institute for Data Intensive Engineering and Science](http://idies.jhu.edu/) at [Johns Hopkins University](http://www.jhu.edu/) and is funded by National Science Foundation award ACI-1261715.
# Reproducibility
The `r Biocpkg('recount')` package `r citep(bib[['recount']])` was made possible thanks to:
* R `r citep(bib[['R']])`
* `r Biocpkg('AnnotationDbi')` `r citep(bib[['AnnotationDbi']])`
* `r Biocpkg('BiocParallel')` `r citep(bib[['BiocParallel']])`
* `r Biocpkg('BiocStyle')` `r citep(bib[['BiocStyle']])`
* `r Biocpkg('derfinder')` `r citep(bib[['derfinder']])`
* `r CRANpkg('devtools')` `r citep(bib[['devtools']])`
* `r CRANpkg('downloader')` `r citep(bib[['downloader']])`
* `r Biocpkg('EnsDb.Hsapiens.v79')` `r citep(bib[['EnsDb.Hsapiens.v79']])`
* `r Biocpkg('GEOquery')` `r citep(bib[['GEOquery']])`
* `r Biocpkg('GenomeInfoDb')` `r citep(bib[['GenomeInfoDb']])`
* `r Biocpkg('GenomicFeatures')` `r citep(bib[['GenomicFeatures']])`
* `r Biocpkg('GenomicRanges')` `r citep(bib[['GenomicRanges']])`
* `r CRANpkg('knitcitations')` `r citep(bib[['knitcitations']])`
* `r CRANpkg('knitr')` `r citep(bib[['knitr']])`
* `r Biocpkg('org.Hs.eg.db')` `r citep(bib[['org.Hs.eg.db']])`
* `r CRANpkg('RCurl')` `r citep(bib[['RCurl']])`
* `r CRANpkg('rentrez')` `r citep(bib[['rentrez']])`
* `r Biocpkg('rtracklayer')` `r citep(bib[['rtracklayer']])`
* `r CRANpkg('rmarkdown')` `r citep(bib[['rmarkdown']])`
* `r Biocpkg('S4Vectors')` `r citep(bib[['S4Vectors']])`
* `r Biocpkg('SummarizedExperiment')` `r citep(bib[['SummarizedExperiment']])`
* `r CRANpkg('testthat')` `r citep(bib[['testthat']])`
Code for creating the vignette
```{r createVignette, eval=FALSE}
## Create the vignette
library('rmarkdown')
system.time(render('recount-quickstart.Rmd', 'BiocStyle::html_document'))
## Extract the R code
library('knitr')
knit('recount-quickstart.Rmd', tangle = TRUE)
```
```{r createVignette2}
## Clean up
file.remove('quickstartRef.bib')
```
Date the vignette was generated.
```{r reproduce1, echo=FALSE}
## Date the vignette was generated
Sys.time()
```
Wallclock time spent generating the vignette.
```{r reproduce2, echo=FALSE}
## Processing time in seconds
totalTime <- diff(c(startTime, Sys.time()))
round(totalTime, digits=3)
```
`R` session information.
```{r reproduce3, echo=FALSE}
## Session info
library('devtools')
options(width = 120)
session_info()
```
```{r 'datasetup', echo = FALSE, eval = FALSE}
## Code for re-creating the data distributed in this package
## Genes/exons
library('recount')
recount_both <- reproduce_ranges('both', db = 'Gencode.v25')
recount_exons <- recount_both$exon
save(recount_exons, file = '../data/recount_exons.RData', compress = 'xz')
recount_genes <- recount_both$gene
save(recount_genes, file = '../data/recount_genes.RData', compress = 'xz')
## URL table
load('../../recount-website/fileinfo/upload_table.Rdata')
recount_url <- upload_table
## Fake urls for now
is.bw <- grepl('[.]bw$', recount_url$file_name)
recount_url$url <- NA
recount_url$url[!is.bw] <- paste0('http://duffel.rail.bio/recount/',
recount_url$project[!is.bw], '/', recount_url$file_name[!is.bw])
recount_url$url[is.bw] <- paste0('http://duffel.rail.bio/recount/',
recount_url$project[is.bw], '/bw/', recount_url$file_name[is.bw])
save(recount_url, file = '../data/recount_url.RData', compress = 'xz')
## Abstract info
load('../../recount-website/website/meta_web.Rdata')
recount_abstract <- meta_web[, c('number_samples', 'species', 'abstract')]
recount_abstract$project <- gsub('.*">|', '', meta_web$accession)
Encoding(recount_abstract$abstract) <- 'latin1'
recount_abstract$abstract <- iconv(recount_abstract$abstract, 'latin1', 'UTF-8')
save(recount_abstract, file = '../data/recount_abstract.RData',
compress = 'bzip2')
## Example rse_gene file
system('scp e:/dcl01/leek/data/recount-website/rse/rse_sra/SRP009615/rse_gene.Rdata .')
load('rse_gene.Rdata')
rse_gene_SRP009615 <- rse_gene
save(rse_gene_SRP009615, file = '../data/rse_gene_SRP009615.RData',
compress = 'xz')
unlink('rse_gene.Rdata')
```
```{r 'cleanup', echo = FALSE, results = 'hide'}
## Clean up
unlink('SRP009615', recursive = TRUE)
```
# Bibliography
This vignette was generated using `r Biocpkg('BiocStyle')` `r citep(bib[['BiocStyle']])`
with `r CRANpkg('knitr')` `r citep(bib[['knitr']])` and `r CRANpkg('rmarkdown')` `r citep(bib[['rmarkdown']])` running behind the scenes.
Citations made with `r CRANpkg('knitcitations')` `r citep(bib[['knitcitations']])`.
```{r vignetteBiblio, results = 'asis', echo = FALSE, warning = FALSE}
## Print bibliography
bibliography()
```
Several functions in the `r Biocpkg('recount')` package have the `outdir` argument, which you can specify to use the data locally. Try using `expressed_regions()` in _SciServer_ to get started with the following code:
```{r 'ER-sciserver', eval = FALSE}
## Expressed-regions SciServer example
regions <- expressed_regions('SRP009615', 'chrY', cutoff = 5L,
maxClusterGap = 3000L, outdir = file.path(scipath, 'SRP009615'))
regions
```
You can find the R code for a full _SciServer_ demo [here](https://github.com/leekgroup/recount-website/blob/master/sciserver_demo/sciserver_demo.R). You can copy and paste it into a cell and run it all to see how _SciServer compute_ works.
## SciServer help
If you encounter problems when using SciServer please check their [support page](http://www.sciserver.org/support/help/) and the [SciServer compute help section](http://compute.sciserver.org/dashboard/Home/Help).
## Citing _SciServer_
If you use [SciServer](http://www.sciserver.org/) for your published work, please cite it accordingly. _SciServer_ is administered by the [Institute for Data Intensive Engineering and Science](http://idies.jhu.edu/) at [Johns Hopkins University](http://www.jhu.edu/) and is funded by National Science Foundation award ACI-1261715.
# Reproducibility
The `r Biocpkg('recount')` package `r citep(bib[['recount']])` was made possible thanks to:
* R `r citep(bib[['R']])`
* `r Biocpkg('AnnotationDbi')` `r citep(bib[['AnnotationDbi']])`
* `r Biocpkg('BiocParallel')` `r citep(bib[['BiocParallel']])`
* `r Biocpkg('BiocStyle')` `r citep(bib[['BiocStyle']])`
* `r Biocpkg('derfinder')` `r citep(bib[['derfinder']])`
* `r CRANpkg('devtools')` `r citep(bib[['devtools']])`
* `r CRANpkg('downloader')` `r citep(bib[['downloader']])`
* `r Biocpkg('EnsDb.Hsapiens.v79')` `r citep(bib[['EnsDb.Hsapiens.v79']])`
* `r Biocpkg('GEOquery')` `r citep(bib[['GEOquery']])`
* `r Biocpkg('GenomeInfoDb')` `r citep(bib[['GenomeInfoDb']])`
* `r Biocpkg('GenomicFeatures')` `r citep(bib[['GenomicFeatures']])`
* `r Biocpkg('GenomicRanges')` `r citep(bib[['GenomicRanges']])`
* `r CRANpkg('knitcitations')` `r citep(bib[['knitcitations']])`
* `r CRANpkg('knitr')` `r citep(bib[['knitr']])`
* `r Biocpkg('org.Hs.eg.db')` `r citep(bib[['org.Hs.eg.db']])`
* `r CRANpkg('RCurl')` `r citep(bib[['RCurl']])`
* `r CRANpkg('rentrez')` `r citep(bib[['rentrez']])`
* `r Biocpkg('rtracklayer')` `r citep(bib[['rtracklayer']])`
* `r CRANpkg('rmarkdown')` `r citep(bib[['rmarkdown']])`
* `r Biocpkg('S4Vectors')` `r citep(bib[['S4Vectors']])`
* `r Biocpkg('SummarizedExperiment')` `r citep(bib[['SummarizedExperiment']])`
* `r CRANpkg('testthat')` `r citep(bib[['testthat']])`
Code for creating the vignette
```{r createVignette, eval=FALSE}
## Create the vignette
library('rmarkdown')
system.time(render('recount-quickstart.Rmd', 'BiocStyle::html_document'))
## Extract the R code
library('knitr')
knit('recount-quickstart.Rmd', tangle = TRUE)
```
```{r createVignette2}
## Clean up
file.remove('quickstartRef.bib')
```
Date the vignette was generated.
```{r reproduce1, echo=FALSE}
## Date the vignette was generated
Sys.time()
```
Wallclock time spent generating the vignette.
```{r reproduce2, echo=FALSE}
## Processing time in seconds
totalTime <- diff(c(startTime, Sys.time()))
round(totalTime, digits=3)
```
`R` session information.
```{r reproduce3, echo=FALSE}
## Session info
library('devtools')
options(width = 120)
session_info()
```
```{r 'datasetup', echo = FALSE, eval = FALSE}
## Code for re-creating the data distributed in this package
## Genes/exons
library('recount')
recount_both <- reproduce_ranges('both', db = 'Gencode.v25')
recount_exons <- recount_both$exon
save(recount_exons, file = '../data/recount_exons.RData', compress = 'xz')
recount_genes <- recount_both$gene
save(recount_genes, file = '../data/recount_genes.RData', compress = 'xz')
## URL table
load('../../recount-website/fileinfo/upload_table.Rdata')
recount_url <- upload_table
## Fake urls for now
is.bw <- grepl('[.]bw$', recount_url$file_name)
recount_url$url <- NA
recount_url$url[!is.bw] <- paste0('http://duffel.rail.bio/recount/',
recount_url$project[!is.bw], '/', recount_url$file_name[!is.bw])
recount_url$url[is.bw] <- paste0('http://duffel.rail.bio/recount/',
recount_url$project[is.bw], '/bw/', recount_url$file_name[is.bw])
save(recount_url, file = '../data/recount_url.RData', compress = 'xz')
## Abstract info
load('../../recount-website/website/meta_web.Rdata')
recount_abstract <- meta_web[, c('number_samples', 'species', 'abstract')]
recount_abstract$project <- gsub('.*">|', '', meta_web$accession)
Encoding(recount_abstract$abstract) <- 'latin1'
recount_abstract$abstract <- iconv(recount_abstract$abstract, 'latin1', 'UTF-8')
save(recount_abstract, file = '../data/recount_abstract.RData',
compress = 'bzip2')
## Example rse_gene file
system('scp e:/dcl01/leek/data/recount-website/rse/rse_sra/SRP009615/rse_gene.Rdata .')
load('rse_gene.Rdata')
rse_gene_SRP009615 <- rse_gene
save(rse_gene_SRP009615, file = '../data/rse_gene_SRP009615.RData',
compress = 'xz')
unlink('rse_gene.Rdata')
```
```{r 'cleanup', echo = FALSE, results = 'hide'}
## Clean up
unlink('SRP009615', recursive = TRUE)
```
# Bibliography
This vignette was generated using `r Biocpkg('BiocStyle')` `r citep(bib[['BiocStyle']])`
with `r CRANpkg('knitr')` `r citep(bib[['knitr']])` and `r CRANpkg('rmarkdown')` `r citep(bib[['rmarkdown']])` running behind the scenes.
Citations made with `r CRANpkg('knitcitations')` `r citep(bib[['knitcitations']])`.
```{r vignetteBiblio, results = 'asis', echo = FALSE, warning = FALSE}
## Print bibliography
bibliography()
```